3 蛋白生化与生物物理技术
3.1.1 E. coli蛋白表达系统
3.1.2哺乳动物表达系统
3.1.3昆虫表达系统
3.1.4硒代蛋白表达
3.2.1 AKTA使用规则及使用实例
3.2.2蛋白纯化柱子的介绍及清洁方法
3.2.3透析袋的使用
3.2.4浓缩管的使用
3.2.5 TEV蛋白酶的使用
3.2.6金属螯合层析
3.2.7分子筛
3.2.8疏水作用层析
3.2.9离子交换层析
3.2.10 Protein A/G 纯化抗体
3.2.11谷胱甘肽转移酶 (GST) 亲和层析
3.2.12 Fab的蛋白表达
3.2.13植物种子蛋白的提取方案
3.2.14包涵体复性
3.2.15 SDS-PAGE凝胶电泳
3.2.16 GraphPad Prism软件的使用
3.3.1酵母双杂交技术从文库中筛选与蛋白X结合的蛋白
3.3.2噬菌体展示
3.3.3配体指数富集的系统进化技术筛选核酸适配体
3.4.1酵母双杂交 Yeast two hybrid
3.4.2双分子荧光互补Bimolecular fluorescence complementation
3.4.3 Western Blotting
3.4.4免疫共沉淀Co-Immunoprecipitation
3.4.5差示扫描荧光技术Differential Scanning Fluorimetry
3.4.6荧光偏振Fluorescence polarization
3.4.7等温滴定微量热Isothermal Titration Calorimetry
3.4.8表面等离子共振技术Biacore
3.4.9蛋白质交联Crosslink
3.4.10 Native-PAGE
3.4.11静态光散射Static light scattering
3.4.12凝胶阻滞实验EMSA-electrophoretic mobility shift assay
3.5.1蛋白酶活性检测
3.5.2 ATP酶活检测
3.5.3解旋酶活性检测
3.5.4体外转录实验
3.1 蛋白质表达系统
3.1.1 E. coli蛋白表达系统
蛋白的原核表达是指用细菌(例如大肠杆菌)表达外源基因蛋白的一种分子生物学技术。原核表达的优点主要在宿主菌生长较快,易于操作,产量较高等。对于结构生物学这类需要大量蛋白质的学科来说比较适用。
常用的大肠杆菌表达系统大多采用T7 promoter,它可以结合Lac operon和Ara operon的元件,对其控制的基因表达做调控。通常目的蛋白的表达可以通过添加诱导剂来实现,本实验室常用诱导剂为IPTG,但也有用四环素和阿拉伯糖诱导原核表达系统。本实验室常用的表达菌株有BL21(DE3),Rosetta,Codon Plus RIPL等。常用的表达载体有V29H,V28E,V30,V55等。不同载体运用的蛋白表达方法不同,所以应根据不同载体的特点选择合适的表达菌株和诱导剂。更多表达载体的信息详见载体部分。
3.1.1.1 E. coli蛋白表达检测
简介(INTRODUCTION)
原核小规模表达测试是大规模蛋白表达前的预实验,只有小规模测试中能够成功表达目的蛋白的克隆才能用于大规模的蛋白表达。此处以小规模蛋白表达检测为例(大规模蛋白表达类同小规模表达,不同的是前者所用培养基的用量以升为单位)。
材料(MATERIALS)
r 试剂(REAGENTS)
无抗性液体培养基(包含试管装5 mL液体培养基)、含抗生素固体培养基、抗生素(视载体情况而定)、诱导剂(1 M IPTG)、60%甘油(使用前需灭菌)、SDS-Loading、考马斯亮蓝。
实验步骤(PROCEDURE)
1. 第一天:转化重组质粒至表达菌株,37 °C培养箱过夜培养16 h。
2. 第二天:挑单克隆于5 mL抗性培养基(一般一种菌株挑取2-5个单克隆),37 °C摇床培养至OD600 =1.2(大约5小时)。
1) 保菌:在超净台台中取菌液和甘油相同体积于冻存管(使甘油终浓度在20%~30%之间,例如0.5 mL菌液+0.5 mL 60%甘油),混匀后放入-80 °C冰箱中保存。
2) 对照:从试管中分别吸取1 mL菌液至2 mL离心管中,不加诱导剂,16 °C摇床中诱导约3小时。
3) 诱导:从试管中分别吸取1 mL菌液至2 mL离心管中,加入适量诱导剂(常为1 μL 1 M IPTG,其他诱导剂用量请参见其使用说明书),16 °C摇床中诱导约3小时。
3. SDS-PAGE检测
1) 诱导组和对照组的菌液,5000 rpm离心5 min,弃上清(此时可以打开煮样器,让其升温)。
2) 每管加入100 μL ddH2O重悬,并加入SDS-loading至1×,混匀,煮样器中煮10~20 min。
3) 12000 rpm离心10 min(此时可以将蛋白电泳装置准备好)。
4) 将离心好的样品取上清按照未诱导、诱导、未诱的方式对应跑胶。
5) 对成功诱导的克隆进行-80 °C冰箱甘油保存。
 |
针对性建议(TROUBLESHOOTING)
1. 尽量不要在冰箱中冻存过多的表达/克隆菌,节约公共资源。
2. 实验室常用的抗生素有Kan和Amp,具体抗性视载体情况而定,例如V28E、V29H等载体抗Kan,V55、V137、V138等载体抗Amp;并不是所有载体都用IPTG诱导,使用前需要查看相关说明书。
3. 若重复若干次实验,目的蛋白的小规模表达均不理想,可以考虑更换载体或表达菌株再进行小规模诱导检测;
4. 若想确定蛋白表达是在包涵体中还是上清,可以从诱导成功的冻存菌中再接种表达菌进行诱导表达,诱导结束后离心菌液取沉淀,加入缓冲液后进行超声破碎,再次离心分别取上清液和沉淀制样,通过SDS-PAGE检测以确定表达的情况。
5. 对于一些特殊性质或较难表达的蛋白,可尝试不同的表达菌株,例如AI可用于表达毒性蛋白,Rosetta、RIP、RIPL常用于表达哺乳动物蛋白(含有稀有密码子)。
3.1.1.2 E. coli大规模蛋白表达
简介(INTRODUCTION)
蛋白质是行使用生物学功能的最重要的一类分子。利用生物化学、生物物理,特别是结构生物学等手段对蛋白质进行研究,需要大量的高纯度的蛋白。利用异源表达宿主来表达重组蛋白是目前最常用的一种方式。
蛋白表达纯化系统有很多种,比较常用的有原核表达系统(大肠杆菌)和真核表达系统(如酵母、昆虫细胞或者哺乳动物细胞)。不同的表达系统各有优缺点,而不同的蛋白对表达系统的要求也不同。这里仅介绍大肠杆菌表达系统。
材料(MATERIALS)
r 试剂(REAGENTS)
LB培养基、抗生素(根据载体抗性而定)、1 M IPTG、50%葡萄糖、2 M MgSO4
r 实验前准备(SETUP)
清洗2 L的摇菌烧瓶;清洗若干个50 mL离心管(灭菌后用于接菌)
r 器械(EQUIPMENT)
摇床、超净台、高压灭菌锅
实验步骤(PROCEDURE)
1. 配制LB:在大规模摇菌前一天配制LB,以5升LB培养基为例:称取50 g NaCl、50 g 蛋白胨、25 g 酵母粉溶于自来水定容至5 L。同时配制用于第二天大规模接菌的LB培养基:称取2.5 g LURIA BROTH(Miller’s LB Broth)溶于100 mL纯水的烧瓶中。将上述LB培养基、50 mL干净离心管、50%葡萄糖、2 M MgSO4于高压灭菌锅121 °C灭菌20 min。
2. 在超净工作台中将小规模诱导成功的表达菌接种于100 mL LB培养基中,并加入相应的抗生素,同时也可每升可添加1 ml 2M MgSO4和5 ml 50%无菌葡萄糖(促进细胞生长,抑制基础/漏表达),37 °C摇床内过夜摇菌。
3. UV灭菌:在第二天大规模摇菌前打开大摇床的UV照射灭菌,同时将LB、灭菌后离心管、抗生素及所需枪头喷洒酒精后放入超净台内,打开UV照射灭菌20 min。
4. 接菌:用无菌50 mL离心管将过夜培养的菌液平分至5升LB中,并加入相应抗生素。
5. 摇菌:37 °C摇床内大规模摇菌至OD600值为1.2(约需3~4小时),然后调节温度至16 °C,加IPTG诱导表达4 h后于4 °C落地离心机收菌,-80 °C冷藏。
针对性建议(TROUBLESHOOTING)
1. 尽量不要使用甘油冻存菌直接接菌,建议重新划线LB平板然后挑取单克隆,以避免噬菌体污染和质粒丢失;
2. 根据蛋白表达量确定大规模摇菌体积,如果表达量很高的蛋白一般3 L足够,而表达量低的蛋白可多摇至10或12 L,一般情况下,我们需要纯化得到几毫克的蛋白用于结晶及后续相关实验用。
3. 由于大摇床降温速度较慢,提前开始降温至16 °C,冷却1 h后再加入IPTG;诱导时间一般4小时足够,具体诱导时间长短,以获得最终有活性非聚合蛋白的量为准,诱导时间过长可能包涵体或聚合体产量大,也有可能发生蛋白降解。
4. 收菌前吸取300 μL菌液制备SDS-PAGE样品,纯化之前先鉴定蛋白表达与否。
5. 诱导的条件,比如时间和温度,可以根据需要进一步优化。
3.1.1.3蛋白纯化
简介(INTRODUCTION)
蛋白质通过原核或者真核表达系统表达后是一种既有宿主蛋白也有目的蛋白的混合体,我们需要通过目的蛋白的特点去进行特异的筛选,把目的蛋白从混合体中分离出来。目前用的较多的一种方法就是金属离子亲和层析(也叫IMAC)的方法。在设计克隆时人为地在目的蛋白的N端或者C端加入6个或者更多连续的组氨酸,组氨酸可以与Ni2+,Cu2+,Co2+结合,再用高浓度的咪唑将目的蛋白洗脱下来。
因此目前市场上常见的IMAC柱有三种:Ni柱,Cu柱以及Co柱。用于螯合这些金属离子的螯合剂有NTA(nitrilotriacetic acid,次氮基三乙酸)和IDA(iminodiacetic acid,亚氨基二乙酸)。所以我们实验室常用的Ni-NTA就是用NTA作为螯合剂的Ni2+离子亲和柱。
三种金属离子柱比较:
| 结合强度 | 特异性 |
Co2+柱 | ★ | ★★★ |
Ni2+柱 | ★★ | ★★ |
Cu2+柱 | ★★★ | ★ |
Co2+柱的结合能力最弱,因此其特异性最好,但损失也是最大的;Cu2+柱的结合能力最强,特异性是最差的,但是产量高,因此对于那些蛋白产量低的情况可以用于初筛;Ni2+柱介于中间。
下面以Ni2+柱为例,介绍IMAC流程。
材料(MATERIALS)
r 试剂(REAGENTS)
Ni-Binding buffer:0.50 M NaCl,16 mM Imidazole,20 mM Tris-HCl pH 8.0
Elution buffer:0.50 M NaCl,400 mM Imidazole,20 mM Tris-HCl pH 8.0
Gel filtration buffer:0.25 M NaCl,10 mM Tris-HCl pH 8.0
r 实验前准备(SETUP)
buffer使用前在放在4 °C冰箱预冷,准备收集管,保证有足够的干净且已烘干的玻璃管
r 器械(EQUIPMENT)
Thermo落地式离心机(使用前预冷至4 °C),AKTA FPLC纯化仪,超声仪,抽滤泵
实验步骤(PROCEDURE)
以MBP融合表达蛋白为例:
A. 纯化蛋白——一次Ni亲和柱
(详见3.2.6金属螯合层析章节)
► 破菌 ►预计消耗时间:30 min~1 h
1. 将冻存在-80 °C冰箱的菌拿出来,放在水里解冻,根据细菌湿重每克5~10 mL加入Ni-Binding buffer。解冻后把金属烧杯放在冰上,进行超声。对于不稳定的蛋白,可加入PMSF以防止蛋白降解。
2. 将烧杯至于冰水中进行超声破碎。液流不呈丝状时,说明已超声完全。
3. 用完后用水洗探头。
▲ 关键步骤:
♫ 观察蛋白质纯化过程中蛋白质的降解和沉淀情况,当蛋白易降解时,可在所有缓冲液及蛋白收集。液中加入通用丝氨酸蛋白酶抑制剂(PMSF,终浓度0.5 mM)抑制蛋白降解。
► 离心、过滤 ►预计消耗时间:40 min
1. 将超声好的菌液平均倒入离心管中,配平好,放入已预冷至4 °C的落地式离心机。
2. 14000 rpm 离心20分钟,将上清倒入小烧杯中。
3. 组装好抽滤装置进行抽滤(注意不要漏,样品和滤瓶均要放在冰上)。
► 平衡Ni-NTA柱 ►预计消耗时间:20 min
1. 将Ni柱从4 °C冰箱取出,这时柱里面是20%酒精。
2. 限压0.4 Mpa,流速3 mL/min,水洗Ni柱10分钟。
3. 换成Ni-Binding buffer再洗10分钟,将Ni柱平衡好。
► 上样使蛋白挂柱 ►预计消耗时间:根据样品体积而定
1. 将Super loop和Ni柱都放在冰上,用洗好的绿色的连接管湿接Ni柱。
2. 蠕动泵流速设定为3 mL/min,限压为0.4 MPa。
3. 当样品流完一个柱体积时,用Ep管接流出的样品,标记好“first flow through”。
4. 当样品快流完时,用Ep管接流出的样品,标记好“last flow through”,用于后续检测。
▲关键步骤:
♫ 样品体积小时可以减速,使样品充分与Ni2+进行反应,能挂柱的都挂上
♫ 第一次纯化某蛋白时,建议收集流出液,避免蛋白不挂柱损失蛋白(注意避免气泡进入柱子)
► 洗脱目的蛋白 ►预计消耗时间:40 min
1. 提前将AKTA的A,B泵用水进行system wash。再用buffer,即A泵用Ni-Binding buffer,B泵用Elution buffer进行system wash。
2. 将系统流速设置1 mL/min,manual run时把Ni柱湿接到AKTA纯化仪上。
3. 调程序(以AKTA Prime为例):设置3 mL/min,0.4 MPa,Load。
check point(mL) | %B | Fraction(mL) |
0 | 4% | 0 |
30 | 4% | 5 |
80 | 60% | 5 |
90 | 60% | 5 |
4. 程序跑完后,将结果保存至自己的文件夹中,写明日期、样品名、程序类型(Ni柱洗脱还是分子筛等)。
5. 在图谱中与目的蛋白的峰对应的样品收集到一个干净的50 mL离心管,取40 μL至Ep管中,标记好“Ni elution”,与前面的样品以及跑蛋白电泳,检测纯化效果。
6. AKTA的A,B泵用水进行system wash。
7. 将用完的收集管用自来水冲5遍,用蒸馏水冲3遍后,晾在试管架上。
8. 用完的Ni柱要进行清洗和重灌镍,详见附录的“柱子型号、特点及清洁方法”。
▲关键步骤:
♫ 注意Ni柱上游的连接管中不要残留不明液体,以免影响纯化效果。
♫ 在多次使用后Ni柱变为白色或灰色时,则需要重新灌填料,以免造成柱子与Ni亲和力差从而影响蛋白纯化。
♫ 柱子使用完毕后清洗并用灌以20%乙醇储存。
► TEV酶切 ►预计消耗时间:12~16 h
(详见3.2.5 TEV蛋白酶的使用)
1. 根据峰图和SDS-PAGE收集目的蛋白,测OD280值。
2. 根据OD280计算并加入相应体积的TEV蛋白酶,同时加入2 mM DTT、1 mM EDTA(终浓度),过夜酶切。
▲关键步骤:
♫ 建议使用TEV和目的蛋白摩尔比例为1:10放置在四度冰箱过夜酶切。
♫ 高盐能够抑制TEV 蛋白酶的活性,因此TEV酶切时尽量控制盐浓度小于0.5M。
♫ TEV消化后,由于DTT和EDTA的原因,需要透析或凝胶过滤才能进行二次Ni纯化。
♫ 在使用TEV酶切蛋白时,需要蛋白透析buffer中存在DTT,因此最好不要一边透析一边酶切,酶切和透析分开来做,这样有助于实现良好的酶切效果。
B. 纯化蛋白——大分子筛
(详见3.2.7 分子筛章节)
► 样品准备 ►预计消耗时间:20 min
1. 实验室有120 mL、320 mL等规格的大分子筛(详见3.2.2 蛋白纯化柱子的介绍及清洁方法),根据洗脱蛋白体积及蛋白性质选择分子筛类型(蛋白洗脱体积要小于分子筛柱体积的1/10)。
2. 倒入高速离心管中,放入已预冷至4 °C的落地式离心机中,14000 rpm离心15分钟。
3. 小心倒入干净的50 mL离心管中,放置在冰上。
4. 用注射器将样品打入Super loop,避免气泡。
5. Super loop连接至AKTA,可以manual run 0.5 mL/min,接好后stop。
▲关键步骤:
♫ 分子筛非常忌讳进气泡,连接时、样品打入Super loop时避免气泡
♫ 过分子筛前样品要离心或者过滤,避免沉淀进入分子筛造成堵塞
► 过分子筛 ►预计消耗时间:4 ~ 5小时
1. 不同的分子量的样品出峰的时间不同,下面以30 mL体积样品,分子量为55 kDa,用AKTA Prime纯化仪来过400 mL柱子为例,设定程序:
check point (mL) | 流速(mL/min) | 限压(MPa) | 连接状态 | Fraction(mL) | %B |
0 | 1.5 | 0.3 | inject | 0 | 0 |
30 | 1.5 | 0.3 | load | 0 | 0 |
250 | 1.5 | 0.3 | load | 5 | 0 |
400 | 1.5 | 0.3 | load | 0 | 0 |
450 | 1.5 | 0.3 | load | 0 | 0 |
▲ 注:程序可以根据实际情况修改
2. 把收集的流出管放在第一个收集管上,分子筛一般会有很多个收集管,如果不从第一管开始的话也可以通过简单计算来确定对应峰的管号。
▲关键步骤:
若是第一次过分子筛,可早些收样,以免错过样品峰
► 收集样品、检测纯度 ►预计消耗时间:1.5小时
1. 将目的蛋白峰所对应的收集管中蛋白倒入干净的50 mL离心管中,放在冰上,并加入5 mM DTT,2 mM EDTA,0.5 mM PMSF(终浓度)。如果要续过二次镍柱,则不需加DTT、EDTA。
2. 取40 μL至Ep管中,标记“gel filtration”来用SDS-PAGE检测样品纯度。
► 分子筛清洗 ►预计消耗时间:8小时
1. 每次用完要用水洗一个柱体积,用20%酒精罐一个柱体积。每用完5次,水洗一个柱体积后,用0.2 M NaOH清洗一个柱体积,再用水清洗3个柱体积,最后用20%酒精罐一个柱体积。详见附录中的“柱子型号、特点及清洁方法”。
2. 清洗完分子筛之后,在标签纸上写明日期、使用者、目前柱子里罐的是什么溶液,方便管理,并且也方便后面的人使用。
▲关键步骤:
♫氢氧化钠要放在塑料瓶中,因为玻璃瓶中的硅酸盐会被碱性的氢氧化钠所腐蚀,成为硅酸钠,因此碱性物质一定不能放在玻璃瓶中。
♫ 清洗及使用分子筛时注意设置限压,不能超过最大压力,否则会破坏分子筛。
C. 纯化蛋白——二次Ni亲和柱
► 平衡Ni-NTA柱(同上) ►预计消耗时间:20 min
► 湿接Ni柱 ►预计消耗时间:10 min
1.提前将AKTA的A,B泵用水进行system wash。再用buffer,即A泵用Ni-Binding buffer(也可用GF buffer),B泵用Elution buffer进行system wash。
2. 将系统流速设置1 mL/min,manual run时把Ni柱湿接到AKTA纯化仪上。
3. 调程序(以AKTA Prime为例):设置3 mL/min,0.4 MPa,Load。
4. 使用A冲洗已接好的Ni柱至UV线齐平。
► 样品准备 ►预计消耗时间:20 min
1. 根据峰图和SDS-PAGE收集目的蛋白,倒入高速离心管中,放入已预冷至4 °C的落地式离心机中,14000 rpm离心15分钟。
2. 小心倒入干净的50 mL离心管中,放置在冰上。
3. 用注射器将样品打入Super loop,避免气泡(上样体积一定要小于柱体积的10%)。
4. Super loop连接至AKTA,可以manual run 3 mL/min,接好后stop。
▲ 关键步骤:
♫ 在纯化的每一步都要进行SDS-PAGE,以鉴定目的蛋白的存在及其纯度。
♫ 连接时、样品打入Super loop时避免气泡。
► 开始程序 ►预计消耗时间:40 min
check point(mL) | %B | Fraction(mL) |
Loop Volume | - | 5 |
20 | 0% | 5 |
80 | 40% | 5 |
90 | 60% | 5 |
▲关键步骤:
大多数蛋白在TEV切掉MBP后不会挂Ni先被洗脱下来,而MBP因含有His tag则会在拉B梯度时才会被洗脱下来,从而达到目的蛋白与MBP分离的目的。不同蛋白性质不同,为保险起见,应从开始程序到结束程序都要进行收集。
有部分蛋白由于其表面有组氨酸和谷氨酸残基簇,可能对Ni柱具有较高亲和力,在洗脱阶段才能被洗脱。这种情况,建议采用gel filtration buffer作为其binding buffer。
在洗脱时建议采用比第一次镍柱更缓和的梯度,便于提高洗脱分辨率。
D. 纯化蛋白—— 24 mL分子筛
► 平衡分子筛 ►预计消耗时间:2~3小时
1. 使用分子筛之前一定要先平衡好,可在泵上或AKTA上进行;注意设定限压和流速,以免破坏分子筛。
2. 先用水冲洗一个柱体积,再用GF buffer平衡分子筛。
3. 将平衡好的分子筛湿接到AKTA上,用GF buffer冲洗分子筛至UV线齐平。
► 开始程序 ►预计消耗时间:30~40 min
1. 将样品4 °C离心机高速离心15 min。
2. 根据样品体积选择合适的loop(0.5/1/2 mL),一般需要选取至少两倍样品体积的loop(比如0.5 mL样品用1 mL loop,1 mL 样品用2 mL loop)。
3. 先用水冲洗三次自动上样环,然后再用GF buffer冲洗3次。
4. 用注射器将蛋白样品打入自动上样环中(注意不要进气泡)。
5. 这里以1 mL loop为例,设定程序:
check point(mL) | Fraction(mL/tube) |
1(loop volume) | 0 |
5 | 0 |
26 | 0.5 |
▲ 关键步骤:
一般24 mL分子筛用于分析蛋白聚合情况,且蛋白质非常容易使分子筛堵塞,尤其是24 mL高分辨率分子筛,上样量要小于10 mg,体积小于1.0 mL。
上样前样品需要4 °C高速离心。
Marker实例:
Bio-Rad蛋白质量标准24 mL分子筛图谱。

► 蛋白保存 ►预计消耗时间:15 min
1. 根据2nd Ni后SDS-PAGE图,来判断接下来是否还需要进一步纯化。如果目的蛋白存在较多杂蛋白,可进一步查阅相关文献了解蛋白性质,然后进一步纯化(疏水柱、离子交换柱等,详见3.2蛋白纯化技术章节);如果目的蛋白条带单一纯度较高,则可测量蛋白浓度并进一步浓缩后于-80 °C冰箱内存放。
2. 蛋白浓度计算公式

▲ 关键步骤:
♫ 蛋白样品采用小体积冷冻(20~200 μL),“速冻速溶”——直接放入- 80 °C冰箱中存放,解冻蛋白时将Ep管握在手中快速解冻,以减少对蛋白的伤害。
♫ OD280测量值一般在0.2~0.8较为准确,若蛋白浓度过高,可稀释后再进行测量。
♫ 测量蛋白质浓度后在保存管上标记清楚。
♫ 将蛋白质浓缩到尽可能高的浓度以便结晶。
♫ 尽量避免经常冻融蛋白质样本。
针对性建议(TROUBLESHOOTING)
1. 蛋白质要尽可能保存在冰上,或者4 °C冰箱中。
2. 提前计划好实验流程,预约摇床、AKTA、平衡分子筛等。
3. 浓缩蛋白质时选择合适的浓缩管,应选择截留小于蛋白质分子量一半的浓缩管。
4. 选用合适的分子筛色谱柱。GE公司的24 mL 10/300 GL 柱型是分析型分子筛,适合对少量蛋白进行分离和鉴定聚合程度。载量建议在5 mg 以内,体积在1 mL以内。Superdex-75适用于5~15 kDa蛋白的分离,而Superdex-200适用于15~100 kDa蛋白的分离,Superdex-300或者Superose-6适用于大于100 kDa的蛋白分离。
5. 为防止蛋白降解,大多数蛋白样品中要添加EDTA和DTT,但是当需要过Ni柱的样品中不能有EDTA和DTT,EDTA是金属螯合剂,它可以把Ni离子从柱子上洗下来,DTT是还原剂,它可以把Ni离子还原成棕色的物质,影响纯化效果。
6. 不同的蛋白质性质不同,在纯化前尽可能多地了解蛋白质性质,包括PI、缓冲体系、潜在的配体,及相互作用蛋白等;一般纯化蛋白用到的buffer的pH值要远离蛋白等电点。
7. 不同的溶解度在pH值、离子强度、温度等方面有很大的不同,当蛋白质在标准条件(如0.25 M NaCl,20 mM Tris-HCl pH 8.0)纯化过程中形成沉淀时,可尝试提高盐浓度、调整pH值、或添加一些稳定剂(2-10%甘油,20-100 mM精氨酸)等方法,试着找到一个易于纯化、结晶的条件。
8. 对于蛋白质交联等无胺缓冲系统,可采用磷酸盐、HEPES等缓冲液。
9. 如果蛋白样品用于圆二色谱实验,需要把缓冲液换成磷酸盐以及低盐体系,降低背景。
10. 如果蛋白样品用于结晶,尽量选用低盐、低缓冲液的buffer,比如纯水,100 mM NaCl,10 mM Tris-HCl pH 8.0。
3.1.2 哺乳动物表达系统
简介(INTRODUCTION)
哺乳动物表达系统相比原核表达等其他系统的优势在于能够使来源于哺乳动物的蛋白质正确的折叠,从而最大程度的接近天然构象,因为该系统能够提供复杂的糖基化等多种翻译后修饰和加工,因而表达产物在分子结构、理化性质和生物学功能方面做接近天然的蛋白质。本实验室目前只是采用最常用的哺乳动物细胞HEK293T和pTT5载体对融合人IgG1 Fc tag的蛋白质进行瞬时表达,并利用Protein A纯化柱对分泌到培养基上清中的蛋白进行纯化,Fc tag可以在纯化后采用TEV酶切除。
材料(MATERIALS)
r 试剂(REAGENTS)
转染试剂PEI:称取0.05 g PEI粉末倒入45 mL超纯水中,利用磁力搅拌。加入12 M盐酸调节pH至小于2,室温搅拌3 h以上至PEI彻底溶解。加入10 M NaOH溶液调节pH至6.9~7.1,加水定容至50 mL。采用0.22 μm针头滤器过滤除菌。分装500 μL/管,-20 °C可以储存1年,解冻后放在4 °C可保存2星期,不能解冻后再复冻。
细胞培养试剂:高糖DMEM、FBS、P/S双抗。
Running buffer:0.15 M NaCl,20 mM Na2HPO4 pH 7.0。
0.1 M乙酸:取2.874 mL冰醋酸加入500 mL ddH2O并混均。
1 M Tris-HCl pH 8.0。
Protein A纯化柱。
r 实验前准备(SETUP)
配置好以上所有试剂,观察CO2是否充足。复苏HEK293T细胞并培养至第二代。抽提无内毒素表达载体质粒。
r 器械(EQUIPMENT)
超净工作台、离心机、蠕动泵、AKTA纯化仪。
实验步骤(PROCEDURE)
1. 培养细胞
1) 提前18~24 h接种适量的HEK293T细胞(约1.5×106/皿)至100 mm培养皿,每个皿加入10 mL DMEM+10% FBS培养。
2) 当细胞融合达到90%左右时,即可进行转染实验。
2. 转染和表达
1) 预热高糖DMEM。
2) 取1根1.5 mL离心管,加入0.5 mL高糖DMEM,再加入8 μg转染的质粒(无内毒素),吹打混匀,命名为溶液1。
3) 取1根1.5 mL离心管,加入0.5 mL高糖DMEM,再加入32 μL PEI试剂(1 mg/mL),吹打混匀,命名为溶液2。
4) 将溶液1加入溶液2,吹打混匀,命名为溶液3。
5) 室温孵育15 min。
6) 滴加1 mL以上溶液3至1皿(悬空逐滴滴到皿的各处),将培养皿前后左右各shake 10次以混匀。
7) 放置培养皿至细胞培养箱培养48~72 h,从培养24 h后开始,每隔12 h收集上清并换液一次。上清中加入终浓度为2 mM EDTA混匀后于4°C储存。
3. 纯化
1) 将收集的培养基上清5000 g、4 °C离心10 min,采用0.45 μm滤膜过滤。
2) 1:2将培养基上清稀释至Running buffer中。测量pH值应为6.0~7.0之间,冰面放置。
3) 使用5 个柱体积的Running buffer平衡Protein A柱子。
4) 将培养基上清稀释液过柱上样,流速设置为4 mL/min。
5) 连接AKTA纯化仪,使用10个C.V.的Running buffer以流速为3 mL/min洗柱,直到OD280达到基线。
6) 使用0.1 M乙酸(pH 3.0~4.5)以3 mL/min流速进行梯度洗脱,梯度从0~100%,洗脱体积为30 mL。每管收集1 mL,每个收集管中加入0.2 mL 1 M Tris(pH 8.0)以中和其酸性。
7) 继续采用4个柱体积的100% 0.1M乙酸洗柱。
8) 用10个柱体积的水洗柱,然后过2个柱体积的20%乙醇,柱子存放于4 °C。
▲ 关键步骤:转染质粒前,细胞的生长状态一定要好。
附:实例结果

从3块150 mm培养皿的HEK293T细胞上清中纯化hTIGIT-TEV-Fc的层析图
针对性建议(TROUBLESHOOTING)
1. 以上步骤中的细胞量及转染试剂量可以按比例缩小或放大。通常一次表达至少需要3个100 mm培养皿。
2. 通常GE公司的1 mL Protein A beads可以结合约20 mg IgG1蛋白。
3. 哺乳动物细胞表达蛋白的量通常较低,不同的蛋白表达量又会不尽相同,如果蛋白表达量高,3个100 mm培养皿的细胞培养上清中可以纯化得到约0.5 mg蛋白。
3.1.3 昆虫表达系统
简介(INTRODUCTION)
昆虫表达系统是一类应用广泛的真核蛋白表达系统,它具有同大多数高等真核生物相似的翻译后修饰加工以及转移外源蛋白的能力。昆虫杆状病毒表达系统是目前国内外十分推崇的真核表达系统。利用杆状病毒结构基因中多角体蛋白的强启动子构建的表达载体,可使很多真核目的基因得到有效甚至高水平的表达。它具有真核表达系统的翻译后加工功能,如二硫键的形成、糖基化及磷酸化等,使重组蛋白在结构和功能上更接近天然蛋白;其最高表达量可达昆虫细胞蛋白总量的50%;可表达非常大的外源性基因(200 kDa);具有在同一个感染昆虫细胞内同时表达多个外源基因的能力。
SF9细胞用于转染、纯化和增殖重组病毒。SF9细胞大小一致,易于操作,能形成良好的单层细胞用于空斑实验。SF9细胞能用于重组蛋白的表达,但是Hi5细胞系的生产量更高。推荐用Hi5细胞系来表达分泌型重组蛋白,其能在无血清培养基中培养,适应悬浮培养以及能生产大量的重组蛋白。
材料(MATERIALS)
r 试剂(REAGENTS)
昆虫培养基、LB、卡那霉素、庆大霉素、四环素
SIM SF培养基(实验室现用):无血清即用型培养基,无需任何添加成分或加血清。然而加2%血清会减少感染过程中蛋白水解量。其优点:1)降低昂贵的血清成本;2)简化分泌重组蛋白纯化过程;3)消除血清组织敏感性。
r 实验前准备(SETUP)
配置液体LB、配置固体LB平板、配制昆虫培养基
r 器械(EQUIPMENT)
Thermo离心机、摇床、培养箱、培养皿、培养瓶、冻存管、移液管
实验步骤(PROCEDURE)
► 制备感受态(DH10Bac)
1. 将DH10Bac加入20 mL LB培养基中(含有50 µg/mL卡那霉素和10 µg/mL四环素)培养过夜,再将培养物接种到500 mL LB培养基(含有卡那霉素和四环素)震荡使OD值达到0.6~0.8。
2. 3000 rpm,4 °C离心15 min,用100 mL 50 mM CaCl2重悬,混合均匀后3000 rpm,4 °C离心15 min。
3. 重复用100 mL 50 mM CaCl2重悬,冰上静置30 min后3000 rpm,4 °C离心15 min。
4. 最后使用20 mL CaCl2 +15% glycerol重悬,分装至无菌的1.5 mL EP管中-80 °C保存。
► 质粒转化
1. 从冰箱中拿出一管100 μL的DH10Bac感受态细胞(DH10Bac感受态细胞本身含有卡那霉素和四环素抗性,而供体质粒pFastbac含有庆大霉素的抗性,所以在制备DH10Bac感受态细胞时需要在培养时添加卡那霉素和四环素,而在制备杆粒时需要使用卡那霉素,四环素,庆大霉素的三抗平板。)加入1~2 µg的质粒混匀,放在冰上静置30 min。
2. 拿入42 °C热激45~90 s,立即放于冰上冷却2 min,加入500 μL无抗性的LB,置于37 °C摇床中225 rpm震荡2~4 h。
3. 涂板(三抗的板子,50 µg/mL卡那霉素,7 µg/mL庆大霉素,10 µg/mL四环霉素,100 µg/mL X-gal,40 µg/mL IPTG),闭光正置于37 °C培养箱中培养至少48 h。
4. 挑选白色的菌落,如果有需要将白色的菌落重新挑到三抗的板子划线,培养至少48 h。
5. 接着挑取白色单克隆至含有5~10 mL的LB(含有7 µg/mL庆大霉素)管中,过夜摇菌后保种放于-80 °C,剩余菌液可以提取Bacmid。
► Bacmid的提取
因为Bacmid大于130 kb,质粒抽提不能使用如上述的膜吸附抽提,而要采用传统的乙醇沉淀法。
1. 准备试剂
Buffer I:25 mM Tris-HCl pH 8.0,50 mM葡萄糖,10 mM EDTA;
Buffer II:0.2 M NaOH,1% SDS(w/v);
Buffer III:3 M KAc,2 M HAc。
2. 将震荡培养的菌液 5 mL离心,弃上清,留下大肠杆菌菌体。
3. 加入250 μL提前加入RNase A 的Buffer I,重悬细菌。
4. 加入250 μL Buffer II,温和充分颠倒4~6次至形成透亮的溶液。
5. 加入350 μL Buffer III,温和颠倒6~8次,13400 rpm离心5~8 min。
6. 将上清转移到洁净灭菌的1.5 mL EP管中,13400 rpm离心5~8 min。
7. 转移上清到洁净灭菌的1.5 mL EP管中,加入等倍体积的异丙醇,冰上放置20 min。
8. 13400 rpm,于4 °C离心10 min,小心移除上清,注意不要吸走白色沉淀或者油状物。
9. 加入1 mL -40 °C预冷的70%乙醇,颠倒混匀后13400 rpm 4 °C离心1~10 min,弃除乙醇,留下白色沉淀,此步骤可重复一次进一步减少杂质。
10. 开盖在干净通风处放置5~20 min至乙醇完成挥发。
11. 加入30 μL预热的ddH2O,充分溶解质粒,测浓度,用于后续实验。
► Bacmid转染(获得P1代病毒)
1. 将准备好的SF9细胞分到一个6孔培养板中,体积为2 mL,密度为1×106,静置15 min让细胞贴壁,轻轻摇动培养板观察到大部分细胞不晃动即可。
2. 取两个1.5 mL EP管分别命名为转染试剂和Bacmid,每管加入100 μL培养基,抽到的Bacmid取4 µg加入到Bacmid管,转染试剂管中加入相应量的转染试剂,静置30 min,再将两管混合孵育30 min。
3. 将混合物滴入到SF9细胞中。
4. 培养皿放于27 °C静置培养5~7 h后更换新鲜培养基继续在27 °C培养66~72 h。
5. 当大多数细胞变圆变肿后收集用1500 rpm, 4 °C避光离心10 min收集上清病毒,可用0.22 µm的滤膜过滤,即获得P1代病毒。
► 病毒感染
1. 同上步,将准备好的SF9细胞分到一个6孔培养板中,体积为2 mL,密度为1×106,但不需要静置使其贴壁。
2. 将P1代病毒以病毒:细胞1:100的比例加入6孔板中,避光在27 °C摇床中培养3~7天。
3. 800 rpm离心5 min取上清,可用0.22 µm的滤膜过滤,即获得P2代病毒。
4. 重复上述步骤扩增病毒即可获得P3、P4代病毒,在扩增获得P4代病毒时,可以扩大细胞体积,采用50 mL细胞,细胞密度不变(若P4代病毒感染细胞表达蛋白水平不理想,可继续扩增)。
5. 获得P1代病毒后,需要对病毒感染细胞表达蛋白的能力进行检测。
6. 将准备好的Hi5细胞分到一个6孔培养板中,体积为2 mL,密度为1×106。
7. 将P1代病毒以病毒:细胞1:100的比例加入6孔板中,避光在27 °C摇床中培养72 h,检测蛋白表达情况;
8. 获得P4代病毒后,可对病毒:细胞比例设计梯度(例如1:10~1:100000)进行蛋白表达量检测,步骤同4,寻找合适比例用于后续大规模蛋白表达。
9. 寻找合适比例后,可进行大规模蛋白表达,Hi5细胞体积可改为50 mL、500 mL、1 L逐步增多,密度同为1×106,按比例加入P4代病毒后,在27 °C摇床中培养3天,即可进行蛋白纯化。
► 分泌蛋白沉淀法
1. 将1 L细胞常温2000 rpm离心15 min,收集1 L的上清,加入沉淀剂,1 mL 1 M NiCl2(终浓度1 mM NiCl2),1 mL 5 M CaCl2(终浓度5 mM CaCl2),50 mL 1 M Tris pH 8.0 (终浓度50 mM Tris pH 8.0)。
2. 加入转子搅拌,会有大量白色沉淀出现。
3. 将这1 L溶液常温6000 rpm离心15 min。
4. 收集上清,0.2 μm滤膜抽滤。
5. 将0.2 μm滤膜抽滤后的上清加入2 mL Nickel beads,室温搅拌3小时以上。
6. 收集Nickel beads,用咪唑洗脱蛋白。
► SF9细胞的冻存
1. 在150×25 mm的培养皿中,25 mL培养基中生长细胞,当细胞密度大于90%时培养基使用1000 rpm离心10 min,然后用6 mL冻存剂重悬(10% DMSO,90% FBS),吸取1 mL放于冻存管中。
2. 将冻存管放于冻存盒,冻存盒依次放于-20 °C冰箱6 h,-80 °C冰箱24 h,液氮罐。
► SF9细胞的复苏
1. 准备SF9培养基:在27~30 °C预热。
2. 从液氮中拿出需要复苏的细胞,迅速放到27 °C水浴中快速摇晃解冻,转移至15 mL离心管中,加入10 mL培养基,1000 rpm离心5分钟,移除上清,加入3 mL培养基,使用移液枪慢慢吹吸三次后转移到60×15 mm培养皿中(依据细胞的量选择培养皿)。
3. 显微镜下检查细胞状态,当大多数细胞贴壁后更换新鲜培养基,将培养皿放到27 °C培养箱中培养至少24 h。
4. 24 h后细胞密度大于80%可以分皿培养,少于80%需要继续更换新鲜培养基培养。
► SF9细胞的传代培养
1. SF9细胞培养在10~15 mL培养基中每代培养2~3天,当密度达到2×106 即可传代,避免超过5×106,影响细胞状态。
2. 预热培养基,室温放置1小时。
3. 准备已培养2~3天的SF9细胞和一个新的锥形瓶,对SF9细胞进行计数,计算其密度。
4. 根据密度吸取一定量的新鲜培养基与SF9细胞至新锥形瓶中,总体积10 mL或可根据需要增大体积,终密度为0.5×106。
► Hi5细胞的传代培养
操作同SF9,不同的是Hi5生长速度较快,每代培养1~2天即可传代。
针对性建议(TROUBLESHOOTING)
1. 冻存(昆虫细胞冻于液氮中,确保细胞处于对数期)。
2. Hi5细胞的冻存与复苏和SF9细胞一样,但50 mL的Hi5细胞可以冻存10~20管。
3. Hi5培养条件是250 mL培养瓶,30~40 mL培养基,在27 °C条件下100 rpm振荡培养,每两天传一次代。
4. 在扩增病毒时,可在SIM SF培养基中加入10%血清。
3.1.4 硒代蛋白表达
简介(INTRODUCTION)
如果某个蛋白没有同源结构,为了确定其相位,需要有重原子衍生物的蛋白晶体。目前最常用的手段就是引入硒代甲硫氨酸(SeMet)的方法。在蛋白表达过程中在培养基中加入硒代甲硫氨酸来替代通常的甲硫氨酸,因此表达出来的蛋白的甲硫氨酸全部取代为硒代甲硫氨酸。我们利用甲硫氨酸缺陷型菌株B834在无机的硒代培养基中表达目的蛋白。B834是一种改造过的表达菌株,缺失甲硫氨酸合成酶,因而不能自身合成甲硫氨酸,但是可以通过氨酰tRNA合成酶将外源的甲硫氨酸或者硒代甲硫氨酸加入到蛋白质的肽链中。
材料(MATERIALS)
r 试剂(REAGENTS)
B834、LB培养基、M9培养基、MediumA培养基、抗生素、硒代甲硫氨酸、IPTG
r 实验前准备(SETUP)
收菌瓶要提前高压灭菌,烘干,预冷。
1× M9 Medium |
17.1 g Na2HPO4.12H2O |
3 g KH2PO4 |
0.5 g NaCl |
1.0 g NH4Cl |
add ddH2O to 1 L |
1× M9 Medium,Glucose,MgSO4,CaCl2 需要高压灭菌;Biotin,Thiamin,antibiotics需用0.22 μm滤膜抽滤。
Medium A: |
1000 mL 1× M9 medium |
8 mL 50% (w/v) glucose |
1 mL 1 M MgSO4 |
0.3 mL 1 M CaCl2 |
1 mL 1 mg/mL Biotin |
1 mL 1 mg/mL Thiamin |
x mL antibiotics |
实验步骤(PROCEDURE)
1. 将重组质粒转化表达菌株B834(DE3)感受态细胞,涂板,37 °C过夜培养。
2. 第二天,挑单克隆到5 mL LB中,37 °C摇过夜。
3. 第三天,将过夜菌全部加到1 L LB中,在37 °C摇床中摇到OD600达到1.0~1.5。
4. 将菌液3700 g,4 °C离心10 min,弃上清,收菌瓶要提前灭菌,并放烘箱烘干,再放4 °C预冷。
5. 加入20 mL Medium A培养基温和重悬菌液,此操作是为了洗去菌液表面残留的LB培养基,使细菌更快适应新的培养基,也避免丰富培养基中甲硫氨酸影响硒代效果。将菌液加入现配的等体积硒代培养基Medium A中(不含任何甲硫氨酸);在37 °C摇床中摇 4~8 h至 OD600=1.5。
6. 加1 mL 50 mg/mL硒代甲硫氨酸,37 °C 生长30 min。
7. 向菌液中加入IPTG诱导, IPTG终浓度为0.25 mM,4 h×37 °C or 20 h×16 °C。
8. 4000 rpm,4 °C,15 min收菌,弃上清。将菌体用样品匙取出,加少量裂解液润洗离心瓶后冻-80 °C冰箱。
针对性建议(TROUBLESHOOTING)
硒代甲硫氨酸价格很贵,因此第一次做,先用甲硫氨酸结合M9培养基试一遍,纯化重组蛋白,计算得率和纯度,摸索所有步骤。然后再用硒代摇2~3 L菌。硒代甲硫氨酸属于有毒物质,请带手套和口罩。
3.2 蛋白纯化技术
3.2.1 AKTA使用规则及使用实例
简介(INTRODUCTION)
FPLC全称为快速蛋白液相色谱(Fast protein liquid chromatography),其原理与高效液相色谱理论类似,是由经典的液体柱层析引入气相色谱理论,并且对相体进行了改革,配用高压输液泵,采用高灵敏检测器、梯度洗脱装置、自动收集装置和微机等发展起来的现代液相色谱。实验室有4套纯化仪,AKTA prime plus,AKTA Pure,AKTA EXPLORER,AKTA FPLC组装机。
AKTA介绍菜单 | ||||
安全(Safety)、组件(Components)、方法编程(Method programming)、使用方法(Usage)、维护注意事项(Maintenance)、针对性意见(Trouble shooting)、使用规则(Rules) |

AKTA Prime Plus纯化系统
► 安全(Safety)
警告!系统必须与接地电源插座连接。
警告!用户不能打开系统,因为系统含高压电路,会造成致命的电击。
警告!必须用上样环或者接线,将上样阀的接口2和6连接。防止转换此阀时,液体从接口喷出,尤其是使用危险化学品时特别危险。
警告!如果有大量溢出的液体渗入系统外壳并与带电零件接触的危险,应立即关闭系统并同指定的技术服务人员联系。
警告!NaOH有害健康,应避免泄漏。
►
|

组件(Components)
AKTA prime plus蛋白质纯化系统包括下列部件:
缓冲液阀和梯度转换阀(Buffer valve and gradient switch valve):缓冲液阀用于选择使用缓冲溶液,用于系统泵施加大的样品体积。梯度转换阀用于建立梯度。
系统泵(System pump):系统泵用于经系统运送液体,如样品或缓冲溶液。将液体经缓冲液阀、梯度转换阀或经上样阀进入流动通道。
压力传感器(Pressure sensor):压力传感器可以测量在位液体压力。还可用作压力保护装置。
混合器(Mixer):混合器用于混合两元梯度。以两步将溶液混合以得到最适宜的结果。混合器的体积可以选择。
上样阀(Injection valve):上样阀用于装加样环和用于将样品注射到柱上。
检测器(Monitor):检测器的目的是测量流出柱后液体的UV吸收、电导率和pH(任选)。用于这些测量的流动池安装在系统的右侧。
具有分流阀的分部收集器(Fraction collector with flow diversion valve):分部收集器用于将样品组分收集在管中供进一步分析。分流阀在废液和收集管之间转换流向。
► 方法编程(Method programming)
主菜单Template | 该菜单出现在自检后启动时,用于运行予制的应用模板和法模板 |
Run stored Method | 用于运行用户编辑的运行方法 |
Manual Run | 用于不使用方法,手工运行系统 |
Program Method | 用于编辑用户专用方法 |
Set Parameters | 用于对检测紫外(UV)、电导、pH、温度、运转泵及混合器设置参数 |
Check | 用于检测系统内部参数,例如序列号、泵运行时间、和灯亮度。 |
利用AKTA Prime Plus 洗脱镍柱中的蛋白(以5 mL镍柱为例)
1. 打开AKTA (机器背部左下角),将A泵和B泵用蒸馏水冲洗干净分别插入到Ni-Binding buffer和Elution buffer中,并在Template中选择system wash,洗泵(大概五分钟)。
2. system wash完毕后,先选择manual run,%B=0,pressure limitation=0.4 MPa,流速=1 mL/min。先将Ni柱底部接到探测器上,再将1号接线口接到镍柱上端(这样可以防止绿色线管扭曲),观察电脑屏幕待UV线、电导线平齐时End program。
3. 设定程序:流速3 mL/min,limit pressure为0.4 MPa
设定breakpoint: |
0 mL:%B=4,set fraction size=0 |
20 mL:开始拉梯度,%B=4;开始收集样品,set fraction size 3 mL/tube |
80 mL:%B=60;同时也一直在收集样品,set fraction size 3 mL/tube |
100 mL:%B=60,set fraction size3 mL/tube |
End |
(意思是:0 mL时所有的系统全部要求置零,0~20 mL区间跑4% Elution buffer把杂蛋白洗下来。20~80 mL区间设定Elution buffer的浓度从4%慢慢升到60%同时收集样品,80~100 mL区间用60%把剩余蛋白全部洗脱下来并收集样品,到达100 mL时停止。)
4. 蛋白纯化结束后,要清洗AKTA泵内部溶液,方便下一位使用者,将A泵和B泵泵头用蒸馏水冲洗干净后放入蒸馏水中,并在Template中选择system wash,洗A、B泵。用蒸馏水清洗完毕后如果长期不用机器,需要用20%酒精再次清洗系统泵。并将收集器上的玻璃管收拾干净,并用水清洗,放于管架晾干。
5. 蛋白质纯化图谱的保存与提取(1)保存:在电脑桌面打开prime view evaluation软件,打来文件夹,在里面按照时间顺序找到你蛋白质纯化图谱,然后点击file,选择save as将你的图保存在你的文件夹。(2)提取:点击file,选择export → curves → 选择01:UV和06:Conc → 并在Reduce number of samples中选择Reduce by 1。点击保存,得到excel 表格,并用Graphpad prism5做蛋白纯化图谱(有视频教学)。
▲ 关键步骤:
♫ 提前看好转盘与滴液体的部分是否靠紧,否则不会自动收集样品。
♫ 根据出峰位置选择样品,一般会有30 mL左右,加入终浓度1 mM EDTA和2 mM DTT以及0.5 mM PMSF,取60 μL制备SDS-PAGE样品。
利用AKTA Prime Plus 过分子筛
1.打开AKTA,将A泵用gel filtration buffer清洗(同上文镍柱清洗)。并用gel filtration buffer手动或者程序设定清洗上样环。
2. system wash完毕后,先选择manual run,%B=0,pressure limitation=0.4 MPa,流速=2 mL/min(根据不同分子筛设定流速)。先将AKTA上1号接线口湿接上部分子筛,此时观察柱子管道是否有气泡,如果有气泡那么应该将1号接口线湿接到分子筛尾部接口,待分子筛头部管道空气排清后再正过来接柱子,最后将柱子尾部接口接到探测器上。观察电脑屏幕待UV线、电导线平齐时End program。
3. Super loop接上,6号出口线接super loop上面,下面出来接上2号口。
4. 设定程序(以30 mL样品和320 mL分子筛为例):
程序 |
流速:2 mL/min,限压:0.4 MPa,A为gel filtration buffer,全程%B=0 |
0 mL:inject |
30 mL:load |
100 mL:load,set fraction size=8 mL/tube(此处的break point体积以各自的蛋白大小而定) |
400 mL:load,set fraction size=0 |
End |
5. 蛋白纯化结束后,要清洗AKTA泵内部溶液,方便下位同学使用,将A泵用蒸馏水冲洗干净插入到蒸馏水中,并在Template中选择system wash,洗泵。
6. 蛋白质纯化图谱的保存与提取:同上文镍柱部分。
♫ 分子筛应当在使用前处理干净并且用对应的buffer平衡至少1.5个柱床体积。
♫ 柱子连接纯化仪时,应该尽量敢走柱子进样管中的气泡(可以通过反向连接柱子,赶走进样管的气泡)。
♫ 接Super loop时先将上端连接inject valve 2号孔后,在manual run中设pathway 为inject,待super loop下端绿色连接管中气泡被赶出后,再接入inject valve 6号孔。
♫ Super loop中的样品如果只有A mL,在设定程序时需要有所保留设定为A-1 mL,以免气泡进到柱子里。
 |
ÄKTA™ pure 是一台灵活直观的层析系统,可用于快速纯化从微克到克水平的蛋白、肽和核酸等目标产物。同时ÄKTA™pure也是一台可靠的系统,它的硬件以及Unicorn软件与各种层析柱和填料仪器可满足任何纯化挑战。系统支持各种层析技术,并满足需要提供最高纯度的自动化要求。系统配置灵活,并可以根据您的需要随时升级,进一步提高其性能。
► 方法编程(Method programming)
主菜单 | 功能介绍 |
Evaluation classic | 用于保存程序和提取数据 |
System control | 运行用户编辑程序 |
Administration | 管理设置参数。(很少使用) |
Method Editor | 用于用户编辑运行方法 |
Method Editor
1. 在file中打开new,会出现Method setting, Equilibration, Sample Application, Elution。在Method setting中主要设置一些基本参数,如柱子类型,体积,流速,压力,Method Bass Unit等。
2. Equilibration这一选项主要是用Buffer平衡柱子,在the total volume is__可以选择平衡柱子的体积。把里面一些杂蛋白洗掉。
3. Sample Application可以选择Super loop体积大小(我们实验室有50和150 mL的Superloop)和Sample volume上样体积,并且可以改变InIet B的百分比。
4. Elution中的Gradient elution在Type中选择linear,并选择target %B和length(mL)。程序设定结束后,点击保存。可以保存在System Control。
|

System control
1. 在System Control中选择Manual用于清洗纯化仪内部管道及泵,具体操作分别点击Pumb A wash和Pumb B wash,选择On,再点击Execute。
2. 在System Control中选择你保存的程序,点击Start.程序就可以运行了,但是在运行前要先清洗泵。程序运行完毕后,点击保存,程序会保存在Evaluation classic。
Evaluation classic
1. 在Evaluation Classic中找你运行后的纯化图谱,保存和提取。
|
![FX6XA(QPM`]7$(QPASEREXO](/_upload/article/images/5d/e1/9aa84513417695bccdbfa06505b3/7ae6bb3a-976b-466f-88ce-67438a41d811.jpg)
2. 在Evaluation Classic中提取纯化图谱,点击File--Curves--Cond and UV,并选择Select,Export。
|

AKTA explorer纯化系统


ÄKTA explorer于2010年12月31日停产,并由ÄKTA avant取代。ÄKTA explorer已广泛应用于行业中,用于快速,稳健的方法和工艺开发。其多功能性和可靠的操作使其成为参与开发纯化过程的实验室的最佳选择。
► 方法编程(Method programming)
主菜单 | 功能介绍 |
Evaluation classic | 用于保存程序和提取数据 |
System control | 运行用户编辑程序 |
Administration | 管理设置参数。(很少使用) |
Method Editor | 用于用户编辑运行方法 |
Method Editor
1. 一般拷贝已有的程序,然后根据自己的需要进行更改。打开文件夹,选择需要拷贝的程序,右键copy,然后选择自己的文件夹,点击OK,拷贝成功。
2. 编辑完程序之后,点击下图箭头所指示标志,选择Gradient检测线性图是否是自己所需程序。
System control
1. 在System Control中选择Manual用于清洗纯化仪内部管道及泵,具体操作分别点击Pumb B wash和Pumb A wash,选择On,再点击Execute。
2. 在System Control中选择你保存的程序,并选择保存路径,点击Start,程序就可以运行了,但是在运行前要先清洗泵。程序运行完毕后,程序会保存在Evaluation classic。
Evaluation classic
1. 在Evaluation Classic中找你运行后的纯化图谱,保存和提取。数据在拷贝后,会出现部分数据丢失的现象,若用于文章作图,建议选择其他纯化仪。
2. 在Evaluation Classic中提取纯化图谱,点击File—Export—Curve,选择UV1_280 and Conc, Reduce by factor调到1,Select到Curves to export, Export.文件类型选择.xls。
► 维护注意事项(Maintenance )
1. 除了连续使用外,当实验完成系统使用结束后,应尽可能避免保存在装载缓冲液状态过夜,尤其高盐浓度洗脱缓!如不能避免,则紧记次日尽早用蒸馏水将系统彻底清洗,再予以保存或再更新使用其它缓冲液,再次投入使用进行实验操作。缓冲液不仅会对系统造成腐蚀,而且高盐浓度缓冲液保存过久造成盐结晶,堵塞管道,损害系统。
2. 若数天或更长的时间不使用系统,除用蒸馏水彻底清洗系统外,取下柱子,换成管道。然后用20%乙醇再冲洗所有使用的管件通道和所有的流动池一遍,并以此将系统保存。
3. 每月定期保养处理,或当发现假峰等问题出现时,可拆下柱子,并用适宜的管道替换其位置,将所有的缓冲液入口管件置于1 M NaOH中,对所有的入口管件通路运行System Wash Method,以流速为1 mL/min,将整个系统充分冲洗20分钟。然后立即用蒸馏水,将整个系统运行System Wash Method加以冲洗20分钟。
4. 一定要保持实验环境的清洁,当使用仪器时发现缓冲液的泄露或看见机器各部件外表有盐的结晶应即刻用清水浸湿的抹布擦拭去除,以免泄漏的液体等造成仪器光学及电子元件的损坏。
5. 当装配部分收集器出口管架时,应根据收集管的长度不同而使用不同的切换口。调整时可将出口管和管夹放在输送臂上的长度导向小孔上,将管夹抵在孔外臂上,将出口管轻推向下到达导向孔的底部,然后上紧管夹螺母。此可确保出口管露出正确的长度,以免造成跳管或收集体积错误情况发生。
6. 微量上样环上样完毕,样品进柱后,应及时注入清水清洗上样环以免蛋白质或高盐堵塞微量上样口及上样环。
7.长期不使用或者其他原因,FPLC 的泵及管路可能有空气,表现为流速不准或没有液体,压力偏低。这时应该把流速调低,接管子用注射器把空气抽出来,prime和FPLC接法不同,可以查看说明书。如果B泵进气,换成B泵,再抽空气。
针对性建议(TROUBLESHOOTING)
1. AKTA每个程序跑完一定要尽快把结果另存了,不然会很快被后面的一大堆数据淹没。每个人建自己文件夹。每个蛋白另建文件夹。
2. 事先做准备工作,比如准备柱子,预约机器,不然无法及时下班。
3. 万一电脑显示器上不更新图谱了,可能是与电脑的连接断了,需要关了纯化仪,重启。
4. 样品收集器偶尔会出现跳管子的情况,收集样品要注意从头或从尾对一下管子。
5. 及时清理废液(尤其过夜纯化蛋白时,要检查废液是否装满)。
6. 不要用蛮力摆动输送臂。松开固定输送臂按纽,并将臂升高。(如图示)
|

► 使用规则
1. 预约:使用纯化仪之前应在对应的纯化仪预约本上预约后在使用;如果临时不适用应在预约本上划去并告知有需要的同学。
2. 上样孔的维护:无论是用Super loop还是微量上样环,使用完毕后,应立即用纯水清洗整个通路,以去除残留在上样环与上样孔中的样品及buffer,以免长期堆积导致结晶堵塞。
3. 连接管的维护:为了防止连接管堵塞(特别是上样环),每次纯化完毕卸下柱子后,应当将所有用过的连接管用纯水冲洗后,再放起来备用。
4. 收集管清理:每次用完仪器后,应及时回收收集管,清理收集盘,以免影响下一位同学使用。
5. 仪器清理:纯化过程中,如果遇到管道漏液,应及时旋紧接头,用抹布擦去液体,并用湿毛巾再擦一遍,避免仪器零部件腐蚀。
6. 接头的使用:为避免滑丝与折断,连接管上的接头不应旋得过紧。
7. 连接管的存放:如果连接管太长,应将连接管盘成圆形存放,以免连接管被卡而造成折损
8. 仪器使用完毕应马上用清水冲洗整个系统泵,防止盐在管路中结晶。
9. UV灯的维护:仪器使用完毕且系统泵清洗过后,如果后面没有同学使用应将电脑与纯化仪关机
1. 金属螯合亲和色谱,又称固定金属离子亲和色谱,其原理是利用蛋白质表面的一些氨基酸,如组氨酸能与多种过渡金属离子Cu2+,Zn2+,Ni2+,Co2+,Fe3+发生特殊的相互作用,利用这个原理可以把富含组氨酸的蛋白质吸附,从而达到分离的目的。
2. 分子筛(size-exclusion chromatography)是指具有均匀的微孔,其孔径与一般分子大小相当的一类物质。常见分子筛为结晶态的硅酸盐或硅铝酸盐,是由硅氧四面体或铝氧四面体通过氧桥键相连而形成分子尺寸大小(通常为0.3~2 nm)的孔道和空腔体系,因吸附分子大小和形状不同而具有筛分大小不同的流体分子的能力。
3. 离子交换层析(ion exchange chromatography)根据蛋白质表面的电荷的不同分离蛋白,带电荷的蛋白和层析填料上相反电荷之间的可逆相互作用进行分离。然后改变条件,结合的组分分别被洗脱下来。
4. 疏水作用层析(Hydrophobic Interaction Chromatography,HIC)是根据分子表面疏水性差别来分离蛋白质和多肽等生物大分子的一种较为常用的方法。蛋白质和多肽等生物大分子的表面常常暴露着一些疏水性基团,我们把这些疏水性基团称为疏水补丁,疏水补丁可以与疏水性层析介质发生疏水性相互作用而结合。
5. Protein A是一种发现于金黄色葡萄球菌(Staphylococcus aureus)的细胞壁表面蛋白,分子量为42 kDa;Protein G是C型或G型链球菌(Streptococcal bacteria)表达的免疫球蛋白结合蛋白。Protein A和Protein G功能相似,能特异性地与哺乳动物免疫球蛋白(Immunoglobulin,Ig)结合,结合的部位通常为免疫球蛋白的Fc区,但有资料显示Protein A也会和人VH3家族的Fab区结合,而Protein G有时与Fab区也有一定结合。同时,两者对于不同的免疫球蛋白亚类的结合能力有所不同。适当重组改造的Protein A、G与琼脂糖凝胶以一定的方式结合,可用于免疫沉淀或抗体的纯化。
6. GST柱是一种旨在利用谷胱甘肽亲和凝胶的高度选择性结合,通过一步法直接从细胞裂解液中对重组谷胱甘肽-S-转移酶标记蛋白进行纯化的试剂。GST融合蛋白是在温和,非变性的条件下洗脱得到的,可以保证蛋白的抗原性和活性。
几种实验室常用的蛋白纯化柱
Type | Volume |
HisTrapTM FF | 1 mL |
HisTrapTM HP | 5 mL |
HisPrepTM FF | 20 mL |
SuperdexTM 200 10/300 GL | 24 mL |
SuperdexTM 75 10/300 GL | 24 mL |
Superdex TM200 26/600 GL | 320 mL |
Tricorn Source Q15(离子柱) | 2 mL/15 mL |
Phenyl(疏水柱) Protein A/G GST | 5 mL 5 mL/1 mL 5 mL |
► 镍柱简介
1. HisTrapTM FF 基本信息

2. HisTrapTM HP基本信息

3. HisPrepTM FF基本信息

► 镍柱清洁方法
1. 使用镍柱前,先用两个柱体积(column volume,CV)的水洗去柱子里的酒精,再用2 CV的Ni-Binding buffer将柱子中的水换成buffer,连接柱时必须湿接。
2. 使用完毕后,先用2 CV ddH2O清洗,最后用20%酒精保存柱子。当柱子使用多次以后或发现压力增加或者颜色变黑,需要对柱子进行深度清洗和重新灌镍。以下是镍柱的清洗步骤。
Regenerate Ni-NTA resin |
Wash with 3 CV water |
Wash with 100 mM EDTA for 2 CV |
Wash with more than 5 CV water |
Wash with 3-5 CV 0.2 M NaOH |
Wash with more than 5 CV water |
Re-charge the resin with 100 mM NiSO4 for 2 CV and stay for > 3 hours |
Wash the resin with more than 10 CV water |
Wash with 20% ethanol 2 CV |
Stock the resin/column in 20% ethanol in 4 °C |
► 分子筛简介
1. SuperdexTM 200 10/300基本信息

2. SuperdexTM 200G 16/600基本信息

3. SuperdexTM 200 26/600基本信息

4. Superdex TM 200 300 mL 基本信息

5. Superdex TM 75 24 mL 基本信息

6. BioRad Erich TM SEC650 24 mL基本信息

▲ 注意:使用分子筛要加滤膜!注意上样体积和总蛋白量。分析型不超过柱体积5%,制备型不超过柱体积10%。采用环形loop上样时,需选用大于样品体积一倍体积的loop。
► 分子筛清洁方法
1. 分子筛使前先1 CV水洗去酒精,然后冲洗1~2 CV gel filtration buffer,使柱子内充满buffer。接柱时必须湿接。分子筛使完毕后,用2 CV ddH2O冲洗柱子,最后用1~2 CV 20%酒精冲洗柱子,用于保存。在分子筛旁边的登记簿上标明柱子状态,使用者和日期。
2. 分子筛使3~4次以后,因为里面会存有杂质(如变性蛋白),用3~4 CV 0.2 M NaOH清洗柱子,然后再用2 CV ddH2O冲洗,最后2 CV 20%酒精填充柱子,并保存。
► 离子柱(Source Q柱)简介


▲ 注意:使用离子交换柱要加滤膜!
► 离子柱清洁方法
1. 使完毕后,5 CV 2 M NaCl 清洗掉残留蛋白→ 5 CV ddH2O清洗NaCl →2 CV 20%酒精填充柱子,用于保存。
2. 若久后柱子很堵或很脏,使完毕后,2 M NaCl过5 CV→ddH2O过5 CV→5 CV 0.2 M NaOH 冲洗柱子,消化杂物→10 CV ddH2O冲洗NaOH →最后2 CV 20%酒精填充柱子,常温保存。
► 疏水柱(Phenyl)简介
1. HIC 5 mL基本信息

► 疏水柱清洁方法
1.使用完毕后,5 CV的 20 mM Tris清洗残留的蛋白→5 CV的ddH2O清洗Tris→2 CV 20%酒精填充柱子,于保存。
2. 若久后柱子很堵或很脏,使用完毕后,用5 CV 2 M NaCl清洗残留的蛋白→5 CV ddH2O清洗NaCl→5 CV 0.2 M NaOH消化冲洗不掉的内含物→10 CV ddH2O再次冲洗→2 CV 20%酒精填充柱子,用于保存。
► Protein A/G简介
1. Protein A 5 mL基本信息

2. Protein G 1 mL基本信息

► Protein A/G清洁方法
用洗脱缓冲液(0.1 M Glycine-HCl pH 2.7)洗脱十个柱体积,ddH2O洗,最后保存在20%乙醇中,放4 °C冰箱。
► GST 简介

► GST清洁方法
1. 沉淀或变性物质的清洗:先用2倍柱体积6 M盐酸胍清洗,再用5倍柱体积纯化缓冲液PBS平衡柱子。
2. 疏水缔合物质的清洗:先用3~4倍柱体积70%乙醇 (或2倍柱体积去垢剂,如 1% Triton™ X-100) 清洗柱子,再用5倍柱体积纯化缓冲液PBS平衡介质。
► 实验室常用柱子类型统计
柱类型 | 数量(根) | |
分子筛 | SuperdexTM 200, 24 mL | 5 |
SuperdexTM 75, 24 mL | 2 | |
SuperdexTM 200, 380 mL | 2 | |
SuperdexTM 200, 300 mL | 1 | |
SuperdexTM 200, 120 mL | 2 | |
BioRad Erich TM SEC650, 24 mL | 1 | |
离子交换柱 | Source Q15 Max, 8 mL | 1 |
Source Q15 tricorn, 2 mL | 1 | |
Source Q15 tricorn, 1 mL | 1 | |
CM Sepharose Fast Flow, 10 mL | 1 | |
Ni-NTA | HisTrapTM HP, 1 mL,5 mL,20 mL | 若干 |
GST | GST, 5 mL | 2 |
疏水柱 | HIC, 5 mL | 若干 |
Protein A/G | Protein A, 5 mL | 4 |
Protein G, 1mL | 2 | |
3.2.3 透析袋的使用
简介(INTRODUCTION)
透析袋应用:除盐、除少量有机溶剂、除生物小分子杂质、浓缩样品。
透析的动力是扩散压,扩散压是由横跨膜两边的浓度梯度形成的。透析的速度反比于膜的厚度,正比于欲透析的小分子溶质在膜内外两边的浓度梯度,还正比于膜的面积和温度,通常是4 °C透析,升高温度可加快透析速度。
使用步骤(PROCEDURE)
1. 选择合适的透析袋
1) 选择合适的截留分子量
选择截留分子量值约为要保留的大分子分子量的一半,以获得至少90%的保留率。(对于需对几种分子进行分离的应用,分子之间的分子量差至少为5倍,才能完成有效的膜透析。分子量差超过100倍时,可以同时保证高收率和高纯度。)
2) 选择适当的扁平宽度(体积)
透析袋扁平宽度的选择取决于样品体积和透析容量。较小的透析管透析更快;较大的透析管因扩散距离较长透析较慢。为易于使用,建议使用总长(包括闭合夹和顶部空间)为大约10~15 cm的透析袋。
3) 选择适合的透析膜材质
对于球蛋白,相对的结合亲和性由低到高为CE<RC<PVDF。
2. 透析袋使用前处理
实验要求不高的,用沸水煮5至10 min,再用蒸馏水洗净,即可使用。对于即用型透析袋:用去离子水清洗即可使用的(这种透析袋具有特殊的生物技术膜,不含硫化物及金属杂质)。
3.透析过程控制
1) 透析液体积:建议为样品体积量的100倍。
2) 透析时间:根据需求。通常允许过夜透析。在持续透析的过程中,至少要进行3次全部更换透析液。建议在透析后2~4 h,6~8 h和10~14 h更换透析液。最后换液后至少继续进行2小时的透析。
▲ 注意:对于高浓度污染物,样品可能需要较长的时间透析,透析液需要更频繁的更换。
3) 透析温度:主要取决于样品。温度最高限制主要取决于膜的类型。纤维素透析袋可以承受的温度高达37 °C。再生纤维素透析袋可以承受的温度可高达60 °C。
4. 透析袋浓缩样品
1) 吹干浓缩法:将蛋白溶液装入透析袋内,放在风扇下吹。此法简单,但速度慢,且温度最好不要超过15 °C。
2) 透析袋浓缩法:将蛋白溶液放入透析袋(无透析袋可用玻璃纸代替),把高分子(6000~12000)聚合物如聚乙二醇(碳蜡)、聚乙烯吡咯、烷酮等或蔗糖撒在透析袋外即可。也可将吸水剂配成30%~40%浓度的溶液,将装有蛋白液的透析袋放入即可。吸水剂用过后,可放入温箱中烘干或自然干燥后,仍可再用。
5. 透析袋的存储
1) 在适当的储存条件下,保质期一般在两年以上。干燥的透析袋要在室温或4 °C储存在聚乙烯袋中。未开封的透析袋4 °C储存。
2) 透析袋一旦变湿,应浸泡在溶液中,不要让透析袋干燥。不断干燥会造成孔隙结构不可恢复的倒塌。
3) 使用后的透析袋洗净后可存于4 °C蒸馏水或者30%乙醇中,确保透析袋始终浸没在溶液内。若长时间不用,可加少量NaN3,以防长菌。从此时起,取用透析袋必须戴手套。

1. 选对截留分子量的浓缩管

2. 使用前处理
1) 清洗
A.新的超滤管用buffer清洗后可直接使用。
B. 重复使用的超滤管:
旧浓缩管使用前处理 |
0.1 M的NaOH浸泡1~2 h |
用水清洗 |
用相关溶液(灭菌水或PBS)离心清洗3次 |
即可用于浓缩新的蛋白 |
▲ 清洗过程不可马虎
2) 判断超滤离心管是否失效
取一相同截留的对照管加水离心,根据滤下速度判断是否失效。或者通过测定滤下液的蛋白浓度判断。
3. 使用过程控制
1) 离心转速:
离心机转速一般采用3000 rpm(一般建议为3000 g,最大不能超过4000 g)。速度过高,目的蛋白会离下,导致目的蛋白丢失;速度过低,耗时,工效低;超滤离心管不能高温高压灭菌(也可参照上文表格)。
2) 减少吸附损失:
滤膜会吸附蛋白,最高可达30%;吸尽蛋白后,再用蛋白buffer吹洗管底。注意用枪头(200 μL)伸入超滤管中时,必须防止碰到滤膜,以免戳破滤膜。
4. 回收保存
不常用的超滤离心管的处理:用0.1 M的NaOH溶液浸泡1~2 h,用水清洗,用20%的乙醇浸泡保存,务必保持滤膜润湿,不能长菌。(注:常检查)。
► 浓缩管脱盐

3.2.5 TEV蛋白酶的使用
简介(INTRODUCTION)
TEV Protease是一种在大肠杆菌中重组表达的带His标签(6×His tag)的烟草蚀纹病毒(Tobacco Etch Virus,TEV)的半胱氨酸蛋白酶,能特异性地识别七肽序列Glu-Asn-Leu-Tyr-Phe-Gln-Gly/Ser,并在Gln和Gly/Ser氨基酸残基之间进行酶切,常用于去除融合蛋白的MBP、Glutathione S-transferase (GST)、His或者其它标签的蛋白酶。
材料(MATERIALS)
r 试剂(REAGENTS)
Buffer:Tris-HCl pH 8.0 EGFP蛋白TEV酶
使用步骤(PROCEDURE)
在这里分别用了四种不同的反应条件来测定TEV酶的蛋白酶活,以下逐个进行介绍。
1. EGFP为带有TEV酶切位点的目的蛋白,浓度为0.37 mg/mL,使用TEV和EGFP不同的摩尔比例1:10,1:20,1:40,1:80,1:160,1:320分别放置在冰上4 °C和室温测试TEV酶切活性。
|

2. EGFP为带有TEV酶切位点的目的蛋白,浓度为0.37 mg/ mL,按照TEV和EGFP摩尔比例1:10分别放置在冰上4 °C和室温测试不同时间点的酶切效果,检测TEV酶切活性。
|

针对性建议(TROUBLESHOOTING)
1. 综上四个不同条件的酶切效果,在保证目的蛋白拥有良好状态的情况下,建议使用TEV和目的蛋白摩尔比例为1:10放置在四度冰箱或冰上过夜酶切。
2. 在使用TEV酶切蛋白时,需要蛋白buffer中存在DTT,因此最好不要一边透析一边酶切,酶切和透析分开来做,这样有助于实现良好的酶切效果。
3. TEV加入量计算公式:
TEV(ml)=
OD280:目的蛋白的OD280
ε:目的蛋白的消光系数
M:目的蛋白的分子量(kDa)
V:目的蛋白体积
TEV:28.745 kDa,消光系数为1.121。
简介(INTRODUCTION)
蛋白质通过原核或者真核表达系统表达后是一种既有宿主蛋白也有目的蛋白的混合体,我们需要通过目的蛋白的特点去进行特异的筛选,把目的蛋白从混合体中分离出来。目前用的较多的一种方法就是金属离子亲和层析(也叫IMAC)的方法。在设计克隆时人为地在目的蛋白的N端或者C端加入6个连续的组氨酸,组氨酸可以与Ni2+,Cu2+,Co2+结合,再用高浓度的咪唑将目的蛋白洗脱下来。
因此目前市场上常见的IMAC柱有三种:Ni柱、Cu柱以及Co柱。用于螯合这些金属离子的螯合剂有NTA(nitrilotriacetic acid,次氮基三乙酸)和IDA(iminodiacetic acid,亚氨基二乙酸)。所以我们实验室常用的Ni-NTA就是用NTA作为螯合剂的Ni2+离子亲和柱。
三种金属离子柱比较:
| 结合强度 | 特异性 |
Co2+柱 | ★ | ★★★ |
Ni2+柱 | ★★ | ★★ |
Cu2+柱 | ★★★ | ★ |
Co2+柱的结合能力最弱,因此其特异性最好,但损失也是最大的;Cu2+柱的结合能力最强,特异性是最差的,但是产量高,因此对于那些蛋白产量低的情况可以用于初筛;Ni2+柱介于中间。
下面以Ni2+柱为例,介绍IMAC流程。
材料(MATERIALS)
r 试剂(REAGENTS)
Ni-Binding buffer:0.5 M NaCl,16 mM Imidazole,20 mM Tris-HCl pH 8.0
Elution buffer:0.5 M NaCl,400 mM Imidazole,20 mM Tris-HCl pH 8.0
r 实验前准备(SETUP)
buffer使用前在放在4 °C冰箱预冷,准备收集管,保证有足够的干净且已烘干的玻璃管
r 器械(EQUIPMENT)
Thermo落地式离心机(使用前预冷至4 °C)、AKTA FPLC纯化仪、超声仪、抽滤泵
实验步骤(PROCEDURE)
► 破菌 ►预计消耗时间:30 min~1 h
1.将冻在-80 °C冰箱的菌拿出来,放在水里解冻,同时按照5 mL/每克湿菌补加Ni-Binding buffer。待解冻一半时通过磁力搅拌器搅拌加速解冻过程,又可以把沉淀重悬,呈均匀状,保证超声时均匀地破菌。
2. 用水洗超声仪探头,方法是:用超声仪超声水。
3. 解冻后把金属烧杯放在冰水混合物中,进行超声。根据菌的多少自定义程序:
| 功率 | 超声时间 | 停止时间 |
菌量多(20 g以上) | 30~35% | 2 s | 2 s |
菌量少 | 20% | 1 s | 2 s |
4. 每5~10 min停止超声,取出烧杯用勺搅拌,并舀一勺菌液再倒入烧杯,液流不呈丝状时,说明已超声完全。
5. 用水洗探头,方法是:用超声仪超声水。
▲ 关键步骤:
♫ 细菌里是我们要的目的蛋白,因此解冻后全程放在冰上。
♫ 超声过程放热,需在冰浴中进行。破碎时,超声探头在液面下1 cm处。
♫ 不时查看,防止冰融使烧杯下沉。
► 离心、过滤 ►预计消耗时间:40 min
1. 提前把离心管洗好,放在管架上待用。
2. 将超声好的菌液平均倒入离心管中,配平好,放入已预冷至4 °C的落地式离心机。
3. 14000 rpm,4 °C离心20分钟。
4. 将上清倒入干净的烧杯中,并放在冰上。
5. 抽滤时,把收集瓶放在冰上,每次倒少一点,抽完再倒。
6. 将抽滤后的样品,用注射器打入已洗好的Super loop中,放在冰上。
7. 剩余残留样品中取100 μL到Ep管中,标记好“before binding”,用枪头抠一点沉淀至100 μL水中,标记好“inclusion body”,用于最后跑胶检测。
8. 抽滤用的虑瓶洗干净,收好。
▲ 关键步骤:将样品打入Super loop的时候尽量不要把空气打进去。
► 平衡Ni-NTA柱►预计消耗时间:20 min
1.将Ni柱从4 °C冰箱取出,这时柱里面是20%酒精。
2.用水洗Ni柱,方法是:用泵将水注入Ni柱,流速为3 mL/min,限压是0.4 MPa。
3. 洗10分钟。
4. 换成Ni-Binding buffer再洗10分钟,将Ni柱平衡好。
► 上样使蛋白挂柱►预计消耗时间:根据样品体积而定
1. 将Super loop和Ni柱都放在冰上,用洗好的绿色的连接管湿接Ni柱。
2. 检查是否漏。
3. 流速为3 mL/min,限压为0.4 MPa。
4. 当样品流完一个柱体积时,用Ep管接流出的样品,标记好“first flow”。
5. 流出液不需要收集。
6. 当样品快流完时,用Ep管接流出的样品,标记好“last flow”,用于后续检测。
▲ 关键步骤:
♫ 样品体积小时可以减速,使样品充分与Ni2+进行反应,能挂柱的都挂上。
♫ 第一次纯化某蛋白时,建议收集流出液,避免蛋白不挂柱损失蛋白。
► 洗脱目的蛋白►预计消耗时间:40 min
1. 提前将AKTA的A,B泵用水进行system wash。再用buffer,即A泵用Ni-Binding buffer,B泵用Elution buffer进行system wash。
2. 将系统流速设置1 mL/min,manual run时把Ni柱湿接到AKTA纯化仪上。
3. 调程序(以AKTA Prime为例):设置3 mL/min,0.4 MPa,Load
check point(mL) | %B | Fraction(mL) |
0 | 4% | 0 |
30 | 4% | 5 |
80 | 50% | 5 |
90 | 50% | 5 |
4. 程序跑完后,将结果保存至自己的文件夹中,写明日期、样品名、程序类型(Ni柱洗脱还是分子筛等)。
5. 在图谱中与目的蛋白的峰对应的样品收集到一个干净的50 mL离心管,加入5 mM DTT,2 mM EDTA以及0.5 mM PMSF(终浓度)。
6.取40 μL至Ep管中,标记好“Ni elution”,与前面的样品以及跑蛋白电泳,检测纯化效果。
7. AKTA的A,B泵用水进行system wash。
8. 将用完的收集管用自来水冲5遍,用蒸馏水冲3遍后,晾在试管架上。
9. 用完的Ni柱要进行清洗和重灌镍,详见附录的“柱子型号、特点及清洁方法”。
▲ 关键步骤:注意Ni柱上游的连接管中不要残留之前实验所剩下的不明液体,以免影响纯化效果。
附:实例结果
 |
针对性建议(TROUBLESHOOTING)
1.蛋白质要时刻保存在冰上,或者4 °C冰箱中。
2. 需要过Ni柱的样品中不能有EDTA以及DTT,EDTA是金属螯合剂,它可以把Ni离子从柱子上洗下来;DTT是还原剂,它可以把Ni离子还原成棕色的杂质,影响纯化效果。
3. 如果样品极其容易被氧化,那么可以向溶液中加入5 mM的β-ME(beta-mercaptoethanol,β-巯基乙醇)(注意要使用时加,并将瓶口用锡箔纸封好以免挥发),Ni-NTA最大可以承受10 mM的β-ME。其余情况尽量不需要加入还原剂。
4. 如果样品不稳定,易降解,可以往菌液中加入0.5 mM PMSF后再进行超声,随后在buffer中也分别加入0.5 mM的PMSF(注意要使用时加)。
3.2.7 分子筛
简介(INTRODUCTION)
分子筛是根据分子大小分离纯化的方法,因此也叫SEC(size-exclusion chromatography)。其中的填料是一种聚糖,是一种均匀的介质。当大小不同的混合物进入分子筛柱时,大的分子因为其路径短而先出来,而小的分子在柱中的路径长而曲折,因此后出来。
该方法主要用于分离单体和聚合物,聚合物是大家试图避免的东西,而变性胶(SDS-PAGE)无法看出样品
 |
中是否有聚合物。分子筛还有几个用途:包涵体复性(re-folding),更换buffer或者过Ni柱前去掉DTT和EDTA等。
材料(MATERIALS)
r 试剂(REAGENTS)
Gel filtration buffer:0.25 M NaCl,10 mM Tris-HCl pH 8.0,蒸馏水(超声去气泡)
r 实验前准备(SETUP)
平衡分子筛:如果柱子里是20%乙醇,那么先用水洗一个柱体积,再用gel filtration buffer进行平衡。
准备足够的干净的已经烘干好的收集管
r 器械(EQUIPMENT)
Thermo离心机(使用前预冷至4 °C)、金属煮样器、AKTA FPLC纯化仪
实验步骤(PROCEDURE)
► 样品准备 ►预计消耗时间:20 min
1. 分子筛一般都在IMAC之后,而Ni柱洗脱得到的蛋白基本在30 mL左右,是比较高浓度的状态,因此分子筛上样之前一定要先高速离心。
2. 倒入高速离心管中,放入已预冷至4 °C的落地式离心机中,14000 rpm离心15分钟。
3. 小心倒入干净的50 mL离心管中,放置在冰上。
4. 用注射器将样品打入Super loop,避免气泡(上样体积一定要小于柱体积的10%)。
5. Super loop连接至AKTA,可以manual run 0.5 mL/min,接好后stop。
▲ 关键步骤:分子筛非常忌讳进气泡,连接时、样品打入Super loop时避免气泡。
► 过分子筛 ►预计消耗时间:4~5小时
1. 不同的分子量的样品出峰的时间不同,下面以30 mL体积样品,分子量为55 kDa,用AKTA Prime纯化仪来过400 mL柱子为例,设定程序:
check point (mL) | 流速(mL/min) | 限压(MPa) | 连接状态 | Fraction(mL) | %B |
0 | 1.5 | 0.3 | inject | 0 | 0 |
30 | 1.5 | 0.3 | load | 0 | 0 |
250 | 1.5 | 0.3 | load | 5 | 0 |
400 | 1.5 | 0.3 | load | 0 | 0 |
450 | 1.5 | 0.3 | load | 0 | 0 |
▲ 注:程序可以根据实际情况修改
2.把收集的流出管放在第一个收集管上,分子筛一般会有很多个收集管,如果不从第一管开始的话也可以通过简单计算来确定对应峰的管号。
▲关键步骤:若是第一次过分子筛,可早些收样,以免错过样品峰。
► 收集样品、检测纯度 ►预计消耗时间:1.5小时
1. 将目的蛋白峰所对应的收集管中蛋白倒入干净的50 mL离心管中,放在冰上,并加入5 mM DTT,2 mM EDTA,0.5 mM PMSF(终浓度)。如果要续过二次镍柱,则不需加DTT、EDTA。
2. 取40 μL至Ep管中,标记“gel filtration”来用SDS-PAGE检测样品纯度。
► 分子筛清洗 ►预计消耗时间:8小时
1. 如果用完分子筛不及时清洗柱,会残留一些蛋白杂质在上面,久而久之会堵塞分子筛。因此每次用完要用水洗一个柱体积,用20%酒精罐一个柱体积。每用完5次,水洗一个柱体积后,用0.2 M NaOH清洗一个柱体积,再用水清洗3个柱体积,最后用20%酒精罐一个柱体积。详见附录中的“柱子型号、特点及清洁方法”。
2. 清洗完分子筛之后,在标签纸上写明日期、使用者、目前柱子里罐的是什么溶液,方便管理,并且也方便后面的人使用。
▲关键步骤:氢氧化钠要放在塑料瓶中,因为玻璃瓶中的硅酸盐会被碱性的氢氧化钠所腐蚀,成为硅酸钠,因此碱性物质一定不能放在玻璃瓶中。
|

附:
A.标准品
B.实例结果
|

针对性建议(TROUBLESHOOTING)
1. 蛋白质非常容易使分子筛堵塞,尤其是24 mL高分辨率分子筛,上样量要小于5 mg,不然一旦分子筛堵了,实验也就毁了。
2. 柱子一定要及时清洗,养成一个好的实验习惯。
3. 不同蛋白的等电点(PI)不同,gel filtration buffer的pH值要远离蛋白的等电点。
3.2.8 疏水作用层析
简介(INTRODUCTION)
疏水层析的分离是基于蛋白质的疏水性氨基酸与基质的疏水性配基之间的可逆性相互作用。
原理:疏水作用在生物系统中广泛存在。蛋白质分子是一个外部有一亲水层包围、内部有疏水核的具有一水基团裂隙。如果将蛋白质表面的疏水基团暴露于高疏水性的环境里,就可以和固定相上的疏水基团结合。定空间的复杂体系,其表面亲水性很强,但也有一些非极性的疏水基团或疏水区域,同时还存在较多的疏疏水作用层析就是依据生物大分子疏水性的差异实现分离的。
|

在高盐环境下,盐离子剥夺蛋白水化层使蛋白质表面的疏水区域暴露,这样它的疏水部分即可与含疏水表面基团的固定相发生较强的疏水相互作用,从而被结合在固定相表面。而一旦降低流动相的盐浓度,蛋白质表面疏水区域闭合,疏水作用降低,这样就可以实现蛋白质的洗脱。图1为疏水吸附过程示意图。
影响疏水层析的因素:
在进行疏水作用层析的分离过程优化时,主要影响因素主要包括:基质类型、配体类型和取代度、盐浓度和盐的类型、pH 等。
常用的疏水介质类型主要可选用苯基-琼脂糖、丁基-琼脂糖和辛基琼脂糖疏水作用层析介质。取代度是基质上疏水配基的密度,取代度的增加在一开始可以增加介质的吸附容量,但是到了一定程度之后由于生物分子之间的空间位阻,吸附趋于饱和。盐在疏水作用层析中至关重要,各种加入到缓冲液和样品溶液的盐都会促进 HIC 中配体和蛋白质相互作用。当这些盐的浓度增加时,被结合的蛋白质量也线性地增加,直到一个特定的浓度,随后在更高浓度下,将以指数形式增加(见图2)。常用的盐类中,进行蛋白质沉淀的强弱或增加水溶液表面张力大小的能力(参考Hofmeister系列排序)如下:Na2SO4 > K2SO4 >(NH4)2SO4 > Na2HPO4 > NaCl > LiCl > KSCN。pH对HIC的影响比较复杂,一般pH的增加会减弱疏水作用,可能是由于增加了带电基团、增加了蛋白质的亲水性所引起的。另一方面,pH的减少能增加表观疏水作用。因此在中性pH值下不键合到 HIC 吸附剂的蛋白质会在酸性的pH下键合上去。
同分子筛相比,疏水柱不限制上样体积。
材料(MATERIALS)
r 试剂(REAGENTS)
Buffer A:1 M (NH4)2SO4,20 mM Tris pH 8.0(注意:Buffer A的盐成分及浓度要依据盐处理过的蛋白样品中的最终盐环境配置)
Buffer B:20 mM Tris pH 8.0
r 器械(EQUIPMENT)
HIC 柱、AKTA 纯化仪、低温高速离心机、电泳槽、电泳仪
实验步骤(PROCEDURE)
1.蛋白质样品处理:
1) 稳定性探究
取少量蛋白样品液作为实验对象,按样品体积将蛋白样中加入高浓度(NH4)2SO4储液或粉末至终浓度为0.5 M,缓慢倒置试管使铵盐溶解,如果此时无蛋白沉淀现象,按上述步骤提高样品的铵盐浓度,依据“加盐→溶解→加盐→再溶解”的程序每次提高铵盐浓度0.1 M或更高,调终浓度为1 M。(如果蛋白在0.8 M以下铵盐浓度时有大量絮状沉淀,则说明该蛋白不适宜用疏水层析,可以通过稀释样品再透析的方法溶解蛋白并除去样品中的高盐。)
▲ 关键步骤:
♫ 加盐原则:蛋白无沉淀,样品中铵盐浓度> 0.8 M,且铵盐浓度越高越有助于提高柱分离能力。蛋白沉淀能力和其疏水基团相关,经验分析,容纳铵盐的浓度为分子量小的蛋白>分子量大的蛋白,浓度低的蛋白>浓度高的蛋白。
♫ 按照Hofmeister系列排序,(NH4)2SO4使蛋白沉淀的能力大于NaCl,如果蛋白在较低浓度的铵盐中沉淀,可以尝试换用NaCl或其他盐,但NaCl浓度推荐大于2 M时再用疏水层析。
2) 样品离心:12000 g 4 °C离心20 min,取上清。
2. 配置Buffer A & B
Buffer A: 20 mM Tris pH8.0,1 M (NH4)2SO4(Buffer A和蛋白样品的最终盐浓度一致)
Buffer B: 20 mM Tris pH8.0
3. 疏水柱处理:
取出相应型号的疏水柱,水洗2个柱体积,再Buffer A平衡2个柱体积,冰上待用。
▲关键步骤:如果疏水柱杂质较多,用前先用 0.2 M NaOH 洗,ddH2O,再用 Buffer A 平衡。
4. 上样及洗脱
1) 用Buffer A & B清洗AKTA的A & B泵,取上清上样。上样及洗脱流速:2~3 mL/ min。
2) 程序设置。以4.7 mL 容量的HiScreen Phenyl FF(High Sub)疏水柱为例:
阶段 | 体积 | 洗脱液含量 | 是否收集 |
上样 | X mL | 0% B | No |
清洗 | 6个柱体积 | 0% B | No |
梯度洗脱 | 10个柱体积 | 0%-100% B | Yes |
再次洗脱 | 2个柱体积 | 100% B | Yes |
5. 电泳鉴定
分别收集流穿液、冲洗液及不同洗脱峰位置的样品,SDS-PAGE电泳鉴定目的蛋白。
6. 柱子清洗及保存
纯化结束后,用ddH2O和20%乙醇先后清洗柱子,4~30 °C保存。也可以保存在0.01 M NaOH中。
附:
 |
实例结果
针对性建议(TROUBLESHOOTING)
HIC期间,样品成分在高离子强度的缓冲液(典型的是1~2 M的硫酸铵或者是3 M的NaCl)中结合到填充柱上。高浓度的盐,尤其是硫酸铵,可能会沉淀蛋白质。因此,需要检测将要使用的结合条件下靶蛋白的溶解性。通常采用降低的盐梯度对靶蛋白进行连续梯度或分布梯度洗脱。
3.2.9 离子交换层析
简介(INTRODUCTION)
离子交换层析(Ion Exchange Chromatography简称为IEC)是以离子交换剂为固定相,依据流动相中的组分离子与交换剂上的平衡离子进行可逆交换时的结合力大小的差别而进行分离的一种层析方法。
离子交换层析中,基质是由带有电荷的树脂或纤维素组成。带有正电荷的称之阴离子交换树脂;而带有负电荷的称之阳离子树脂。当蛋白质处于不同的pH条件下,其带电状况也不同,pH大于等电点时蛋白带负电荷。阴离子交换基质结合带有负电荷的蛋白质,所以这类蛋白质被留在柱子上,然后通过提高洗脱液中的盐浓度等措施,将吸附在柱子上的蛋白质洗脱下来。结合较弱的蛋白质首先被洗脱下来。反之阳离子交换基质结合带有正电荷的蛋白质,结合的蛋白可以通过逐步增加洗脱液中的盐浓度或提高洗脱液的pH值洗脱下来。
优点:对于不够纯的样品进行进一步纯化。对没有融合标签的蛋白以及天然蛋白的纯化尤为适用。
材料(MATERIALS)
r 试剂(REAGENTS)
Buffer A: 10 mM Tris pH 8.0
Buffer B: 1 M NaCl,10 mM Tris pH 8.0;20%乙醇
r 实验前准备(SETUP)
干净的样品收集管、冰、高速离心机预冷、SDS-PAGE胶
r 器械(EQUIPMENT)
Q柱、AKTA纯化仪、低温高速离心机、电泳槽、电泳仪、泵
实验步骤(PROCEDURE)
1. 过阴离子交换柱前,采用透析或者过分子筛脱盐处理,将蛋白样品溶液置换为10 mM Tris pH 8.0。样品需要离心除去沉淀。柱子前面接上滤膜使用。
2. 平衡柱子(此处用的是8 mL Source Q柱子),流速为2 mL/min,限压2 MPa。先用Buffer B冲洗柱子残留的杂蛋白,洗到当A280的吸光值回到基线为止,大概十个柱体积。再换成 Buffer A洗十个柱体积。
3. 样品体积大于5 mL用Super Loop上样;若小于5 mL,采用上样环上样。
4. 将样品以2 mL/min上样,5 mL/tube收集。
5. 采用Buffer A冲洗10 CV,5 mL/tube收集。
6. 采用线性梯度0~50% Buffer B共10 CV洗脱蛋白,5 mL/tube收集。50~100% Buffer B共3 CV洗脱蛋白,5 mL/tube收集。
7. 分别收集整个过程中所出现的峰,SDS-PAGE胶鉴定。
8. 纯化结束后,用buffer B冲洗5 CV,再用水冲洗5 CV,最后注入20%乙醇,放4 °C冰箱。
附:
|

实例结果

针对性建议(TROUBLESHOOTING)
1. 明确蛋白的等电点,根据蛋白的带电情况选择阴离子或阳离子柱,预先预测一下目的蛋白是出现在流穿还是在洗脱中出峰。
2. Q柱结合的杂质比较多,如果用Buffer B不能除去杂质,可以用0.2 M NaOH清洗杂蛋白,再依次采用ddH2O、Buffer B和Buffer A过柱。
3. 第一次纯化时,最好流穿和洗脱液都收集,以免蛋白丢失。
4. 以上流程不一定适用于所有蛋白质。还需要多摸索纯化条件比如Buffer的pH,洗脱的最适盐浓度等等。
3.2.10 Protein A/G 纯化抗体
简介(INTRODUCTION)
Protein A 和G是细菌细胞壁的一个构成组分,它们对免疫球蛋白的Fc区域有高度亲和性。以A为例,它包含5个与IgG的Fc段结合的区域,当作为亲和配体配体固定于琼脂糖上时,它的这五个结合区域则空闲出来以捕捉IgG,从而达到富集纯化抗体的目的。Protein G和A与抗体结合方式相同,只是A经过人工改造,添加了C末端Cys用以形成二硫键与琼脂糖结合。Protein G和A对不同抗体亚型具有一定的选择性。Protein A和G凝胶柱可以重复使用。
材料(MATERIALS)
r 试剂(REAGENTS)
结合缓冲液PBS(20 mM PBS、0.5 M NaCl pH 7.0)、洗脱缓冲液(0.1 M Glycine-HCl pH 2.7)、20%乙醇、1 M Tris pH9.0
r 器械(EQUIPMENT)
Protein A/G柱、AKTA纯化仪、低温高速离心机、电泳槽、电泳仪、抽滤泵、抽滤瓶、0.8 μm 滤膜
实验步骤(PROCEDURE)
1. 用至少5个柱体积的0.1 M Glycine-HCl pH 2.7缓冲液再生Protein A柱子。
2. 用5~10个柱体积的结合缓冲液PBS pH 7.0平衡柱子。
3. 将样品上柱,流速要慢,1~2.5 mL/min。
4. 用30个柱体积的结合缓冲液洗脱掉不能结合的杂蛋白。
5. 当280 nm (A280)的吸光值回到基线时,加洗脱缓冲液0.1 M Glycine-HCl pH 2.7。当吸光曲线表明蛋白质开始流出柱子时(此为纯化抗体),在收集管中加入1 M Tris pH 9.0来, 其量为抗体洗脱体积的1/10。
6. 纯化抗体全部完成后,应采用0.01 M PBS pH 7.0溶液对纯化的抗体进行透析,透析体积应为样品体积20倍,透析4小时以上。透析后抗体有可能需要进行浓缩,因为透析可能导致抗体被稀释。大多数抗体的浓度可被安全的浓缩到1~10 mg/mL。
7. 纯化结束后,再生柱子,用结合缓冲液洗脱十个柱体积,ddH2O洗,最后保存在20%乙醇中,放4 °C 冰箱。
▲关键步骤:
♫ 柱子再生很重要,特别是用一个柱子纯化不同抗体时,第一步需要确定柱子上不含有残留的抗体。
♫ 上样时的流速对样品结合效率有影响,不宜过快。
♫ 收集洗脱的抗体后,尽快加入1 M Tris pH 9.0 buffer来中和pH,避免抗体失活,若抗体后续需与CNBr beads则不可使用Tris,可用磷酸二氢钠。
针对性建议(TROUBLESHOOTING)
1. 第一次纯化时,最好流穿和洗脱液都收集,以避免抗体丢失。
2. 以上流程不适用于所有抗体,例如使用何种缓冲液可根据具体实验材料自行摸索调整。
3. 纯化前根据不同样品来源,需要进行相应的样品预处理。例如腹水样品需提前离心,去除絮状脂肪,再用0.22 μm 滤膜抽滤,否则会堵塞柱子。若样品中杂质过多也可先进行硫酸铵沉淀来粗纯样品。样品上样前都需经过离心和0.8 μm 滤膜抽滤。
3.2.11 谷胱甘肽转移酶 (GST) 亲和层析
简介(INTRODUCTION)
谷胱甘肽S转移酶(GST)是一个含有211个氨基酸的蛋白,通常将该蛋白加入到重组蛋白的末端进行蛋白的纯化和检测。与常用的组氨酸标签易受组蛋白的影响产生非特异性结合,从而导致融合蛋白纯度不高不同。
谷胱甘肽转移酶(GST)与其底物-谷胱甘肽是之间具有极高的特异性结合,结合方式为硫键共价结合,所以可以获得更高的纯度。带有GST标签的融合蛋白(V9或V10)与GST琼脂糖凝胶孵育,凝胶手臂上的谷胱甘肽与GST蛋白特异性结合,通过洗脱去除不能与凝胶结合的杂蛋白,进一步使用还原型的谷胱甘肽(GSH)洗脱时,GSH可以竞争GST上的结合位点从而将GST蛋白洗脱下来,从而获得纯化的GST融合蛋白。1 mL树脂可结合5~8 mg融合蛋白。与镍柱层析一样,整个操作过程需要在冷库中进行。细胞破碎过程与镍柱层析一样。
材料(MATERIALS)
r 试剂(REAGENTS)
纯化缓冲液PBS(135 mM NaCl、2.7 mM KCl、1.5 mM KH2PO4、8 mM K2HPO4 pH 7.4)、GSH、1 M DTT、0.5 M EDTA、高pH缓冲液(0.1 M Tris-HCl、0.5 M NaCl pH 8.5)、低pH缓冲液(0.1 M sodium acetate、0.5 M NaCl pH 4.5)、1% Triton™ X-100、70%乙醇
r 实验前准备(SETUP)
干净的样品收集管、冰、菌体破碎后样品需4 °C离心取上清抽滤除杂、SDS-PAGE胶
r 器械(EQUIPMENT)
AKTA纯化仪、低温高速离心机、GST柱、电泳槽、电泳仪、抽滤泵、抽滤瓶、0.8 μm滤膜
实验步骤(PROCEDURE)
1. 蛋白的表达和裂解步骤同蛋白大规模表达步骤。
2. 用10个柱体积的纯化缓冲液PBS平衡GST柱。
3. 然后将蛋白上清加入GST柱中,流速控制在1 mL/min,并收集流穿。
4. 上样结束后,用PBS清洗4个柱体积,收集流穿液。
5. 用PBS现配10 mM GSH,用两个柱体积洗脱,收集流穿液用于下一步纯化,可依据下一步纯化决定是否加DTT、EDTA、蛋白酶抑制剂等以防蛋白降解。
6. 用10倍柱体积的纯化缓冲液PBS重新平衡GST柱子。
7. 各步骤收集的样品进行SDS-PAGE检测。
8. 如果在使用一段时间后,柱子因表面沉积过多杂质导致蛋白结合能力下降,需对柱子进行清洗。步骤见附录。
针对性建议(TROUBLESHOOTING)
1. 第一次纯化时,每步都要留样,跑SDS-PAGE胶,便于分析纯化效果。
2. 因GSH易氧化,需现用现配。
常见问题与解决方案(COMMON PROBLEMS AND SOLUTIONS)
1. 沉淀或变性物质的清洗:
1) 用2倍柱体积6 M盐酸胍清洗。
2) 再用5倍柱体积纯化缓冲液PBS平衡柱子。
2. 疏水缔合物质的清洗:
1) 用3~4倍柱体积70%乙醇(或2倍柱体积去垢剂,如1% Triton™ X-100)清洗柱子。
2) 再用5倍柱体积纯化缓冲液PBS平衡介质。
3. 介质再生:
每次层析前,为达到最佳纯化效果,需对柱子进行再生,步骤如下:
1) 2倍柱体积高pH缓冲液(0.1 M Tris-HCl,0.5 M NaCl pH 8.5)和低pH缓冲液(0.1 M sodium acetate,0.5 M NaCl pH 4.5)交替洗脱三次。
2) 10倍柱体积纯化缓冲液PBS平衡柱子。
3.2.12 Fab的蛋白表达
简介(INTRODUCTION)
Fab是抗体中能直接结合抗原的区域。抗体被木瓜蛋白酶切割后,一般情况是是产生两个Fab片段和一个Fc片段。Fab保留了完整抗体结合抗原的能力,且能够结晶(完整的抗体很难结晶)。原核表达抗体和Fab具有较大难度,主要原因是抗体是分泌蛋白,且具有链间二硫键,在细菌中折叠困难,常以包涵体形式表达。
pFab载体采用细菌phoA(碱性磷酸酶)启动子,其特性是在无磷酸的培养基环境下,宿主细菌(55244)能够活化该启动子,缓慢地将表达的蛋白分泌到细菌周质腔(周质腔的环境能够帮助蛋白折叠正确)。我们将Fab克隆到pFab质粒,能够很好的在55244菌种分泌表达出具有活性的Fab。由于周质腔空间较小,此法的缺点是蛋白获得量较低。
材料(MATERIALS)
r 试剂(REAGENTS)
1. 10×CRAP-Phosphate Media
10×Stock | 1 L | 2 L | 4 L |
(NH4)2SO4 | 35.7 g | 71.4 g | 142.8 g |
NaCitrate-2H2O | 7.1 g | 14.2 g | 28.4 g |
KCl | 10.7 g | 21.4 g | 42.8 g |
Yeast Extract | 53.6 g | 107.2 g | 214.4 g |
Hy-Case SF Casein | 53.6 g | 107.2 g | 214.4 g |
ddH2O | UP to 1 L | UP to 2 L | UP to 4 L(use hot water to save time) |
2. 1×CRAP-Phosphate Media:
配制步骤:确定所需的1xCRAP-Pi体积,确定加入水的体积,加入水的体积为总体积减去10xCRAP的体积减去以下三个溶液的体积,加入计算出体积的水,计算加入MgCl2的质量(1xCRAP-Pi中MgCl2的终浓度为100 mM),并称量后加入。计算加入NH4OH氨水的体积(25%~28%的氨水摩尔浓度为约14 M,1xCRAP-Pi中氨水的终浓度为100 mM)并加入。室温搅拌1 h,NH4 MgPO4·xH2O会析出沉淀,采用滤纸抽滤,去除沉淀。调节pH至7.3。加入下表中的溶液,至此,1xCRAP配制完成。可以立即使用,也可以放置在4 °C冰箱过夜。
1×CRAP-Pi | 1 L | 2 L | 4 L |
1 M MOPS pH 7.3 | 110 mL | 220 mL | 440 mL |
50% glucose | 11 mL | 22 mL | 44 mL |
1 M MgSO4 | 7 mL | 14 mL | 2 mL |
3. 2×YT培养基:1 L培养基含16 g胰蛋白胨,10 g酵母提取物,5 g氯化钠,高压灭菌。
r 实验前准备(SETUP)
制备表达菌株E. coli 55244的感受态
r 器械(EQUIPMENT)
超净工作台、水浴锅、灭菌锅、摇床、烧瓶、Thermo离心机、抗生素、摇床、定性滤
实验步骤(PROCEDURE)
► Fab表达
1. 将构建好的pFab质粒转化表达菌55244感受态100 μL,加入900 μL 2xYT,30 °C,220rp培养1 h。不涂布平板,从新鲜转化菌开始,不使用冻存的菌种。
2. 将以上1 mL转化菌全部接种到10 mL 2×YT中,30 °C,200 rpm培养过夜。
3. 将以上10 mL过夜菌液接种至1 L 2×YT/Amp中(具体情况视最终表达量而定,总之是1:100接种),置于带挡板的摇瓶中,30 °C,200 rpm培养过夜。
4. 离心细菌(4000 rpm,离心10 min),弃上清,使用等体积的1xCARP-Pi/Amp培养基重悬菌体。
5. 将精确的1 L 1xCARP-Pi/Amp培养基细菌悬液置于2 L无挡板摇瓶,用锡箔纸将瓶口封严,使用胶布包裹严实,严防通气,30 °C,230 rpm培养24~27 h。
6. 稀释10×后测量OD600,离心(4000 g,10 min,4°C),弃上清。不要冻存细菌,继续往下做。
7. 采用裂解缓冲液(Protein A 柱纯化用50 mM Tris,150 mM NaCl,2 mM EDTA pH 8.0;镍柱纯化用20 mM Tris,500 mM NaCl,5%甘油,0.1%Tween20 pH 8.0,使用前加入PMSF)重悬菌体(1:8)。
8. 冰浴超声破菌。
9. 离心(17000 g,4 °C,30 min),取上清,采用Protein A 或者镍柱纯化。
► Fab纯化(以下两种方法二选一)
采用protein A柱子纯化
1. 使用3个柱体积(C.V.)的1 M 乙酸过柱,以再生protein A柱子,流速设置为2.5 mL/min。
2. 使用10 个C.V.的running buffer(如破菌液)平衡柱子。
3. 将以上破碎上清过柱,流速设置为1 mL/min。
4. 采用纯化仪,使用10个C.V.的running buffer以流速为1 mL/min洗柱,直到OD280达到基线。
5. 使用50 mL 0.1 M乙酸以1 mL/min进行洗脱,将洗脱液分为5 mL fraction(Fab将在前10 mL洗脱),每个fraction中加入1 mL 1 M Tris。
6. 将Fab透析到PBS中。
7. 重生protein A柱子,并使用大量的水冲洗,采用20%乙醇4 °C储存。
采用镍柱纯化
1. 使用5个柱体积的破菌缓冲液平衡柱子,流速设置为2.5 mL/min。
2. 将以上破碎上清过柱,流速设置为2.5 mL/min。
3. 采用纯化仪,使用25 mL,0~200 mM咪唑进行梯度洗脱,将洗脱液分为5 mL fraction。
4. 将Fab透析到PBS中。
5. 重生柱子,采用20%乙醇4 °C储存。
针对性建议(TROUBLESHOOTING)
1. 接种过程不要污染杂菌。
2. 由于这一表达质粒是持续表达,因此不适宜做甘油冻存。每次都需要从质粒转化开始。
3. Fab偶尔发现有降解发生,需要添加蛋白酶抑制剂,同时尽快纯化速度。
4. 该体系也可用于其他需要分泌的蛋白表达。
3.2.13 植物种子蛋白的提取方案
简介(INTRODUCTION)
植物种子里有大量的蛋白,主要以存储蛋白为主,可以占到总蛋白的70%以上。同时,种子里还有大量纤维,不溶于水。另外,种子里还有大量的脂类,可以用环己烷或正己烷脱脂。
|

根据蛋白分子在传统的蔗糖密度梯度离心里的分布,种子蛋白通常有三大类。他们从大到小分别是2 S、7 S和11 S。这种只是粗略的分类,通常有一种或者多种分子量(聚合物分子量,不是单体分子量)接近的蛋白。2 S(分子量在10~15 kDa)和7 S(单体分子量在60 kDa,三聚体150~210 kDa)一般能溶于低盐溶液,用水萃取。而11 S单体分子量在60 kDa,六聚体360 kDa),也能溶于水,但是更易溶于高盐(1 M NaCl)溶液,因此可以先用低盐,再用高盐萃取。2 S中还有只脂溶性,但是不溶于水的蛋白,也是花生芝麻等食物中重要的过敏原。
材料(MATERIALS)
r 试剂(REAGENTS)
种子20 g~50 g
r 器械(EQUIPMENT)
粉碎机(榨汁机)、离心机(使用前预冷至4 °C)、金属煮样器、AKTA FPLC纯化仪
实验步骤(PROCEDURE)
► 样品准备 ►预计消耗时间:20 min
1. 选取20~50 g新鲜种子去皮去壳,加入200 mL低盐萃取液Buffer A (10 mM Tris-HCl pH 8.0)溶液,用匀浆器破碎。加100 mL正己烷,室温搅拌2~4小时。14000 rpm 离心20分钟,去除上层油脂层,中间水相用纱布,然后用卡纸过滤,再用0. 8 μm 滤膜过滤,得到组分F1,即水提取物。如果蛋白得率很低,可以在萃取液中加少量盐,比如100 mM NaCl。但是注意需要透析后过离子交换柱。
2. 离心沉淀加100 mL高盐萃取液Buffer B (10 mM Tris-HCl,1 M NaCl pH 8.0), 再次用匀浆器搅拌破碎。14000 rpm 离心20分钟,上清用纱布,然后卡纸过滤,再用0.8 μm 滤膜过滤,得到组分F2,即盐提取物。
3. F1用XK 26/60 Superdex-200分子筛过滤。按照出峰位置,分离11 S,7 S和2 S蛋白。分子筛后各组分,进一步用Source Q阴离子交换柱分离蛋白。Buffer A是Binding buffer, Buffer B是Elution buffer。SDS-PAGE 鉴定后,可以进一步过HIC纯化。Binding buffer 需要加1~2 M硫酸铵。
4. F2 组分相对单一,可以直接过Superdex 200分子筛,主要是11 S组分,同时有2 S组分。11 S需要过HIC纯化。Binding buffer 需要加1~2 M 硫酸铵。
▲ 关键步骤:种子不要太多,尽量打碎。脱脂和过滤要充分。
► 预期结果 ►预计消耗时间:4~5小时
下面是以松籽为例,纯化7 S vicilin(pink 1)蛋白的部分结果。


Figure 1Purification of pine nut crude extract by gel filtration. Pine nuts were liquefied in PBS.Crude extracts was filtered, and loaded into a 26/70 Superdex-200 column. Pin k 1 was eluted as a major peak at 190 ml, corresponding to a molecular weight of 126 kD.
Figure 2.Anion exchange purification of Korean pine vicilin. The vicilin containing peak from the gel filtration step was pooled and loaded onto an 8 mL Source 15Q anion exchange column pre-equilibrated with the 10 mM Tris buffer. The column was eluted using the Tris buffer with a linear NaCl gradient (0-0.3 M over 75 mL) by mixing the Tris buffer with buffer B (10 mM Tris, pH 7.9, 1 M NaCl). The vicilin peak is indicated.
针对性建议(TROUBLESHOOTING)
有一些种子存储蛋白,特别是11 S globulins, 其溶解度对盐和温度非常敏感。在高盐溶液中非常可溶,而在低盐或者低温下,溶解度很低。可以利用这一性质进行纯化和结晶。
3.2.14 包涵体复性
简介(INTRODUCTION)
 |
在某些生长条件下,大肠杆菌能积累某种特殊的生物大分子,它们致密地集聚在细胞内,或被膜包裹或形成无膜裸露结构,这种水不溶性的结构称为包涵体。当外源基因在原核细胞中高效表达时,形成的包涵体是由膜包裹的高密度、不溶性颗粒,一般含有50%以上的重组蛋白,其余为核糖体元件、RNA聚合酶、外膜蛋白等,大小为0.5~1 μm,不溶于水,只溶盐酸胍或尿素。
► 包涵体形成原因复杂,主要有以下几点:
1. 基因工程菌的表达产率过高,超过了细菌正常的代谢水平,由于细菌的δ因子的蛋白水解能力达到饱和,使之表达产物积累起来。研究发现在低表达时很少形成包涵体,表达量越高越容易形成包涵体。原因可能是合成速度太快,以至于没有足够的时间进行折叠,二硫键不能正确的配对,过多的蛋白间的非特异性结合,蛋白质无法达到足够的溶解度等。
2. 重组蛋白的氨基酸组成:一般说含硫氨基酸越多越易形成包涵体,而脯氨酸的含量明显与包涵体的形成呈正相关。
3. 重组蛋白所处的环境:发酵温度高或胞内pH接近蛋白的等电点时容易形成包涵体。
4. 重组蛋白是大肠杆菌的异源蛋白,由于缺乏真核生物中翻译后修饰所需酶类和辅助因子,如折叠酶和分子伴侣等,致使中间体大量积累,容易形成包涵体沉淀。
5. 蛋白质在合成之后,于中性pH或接近中性pH的环境下,其本身固有的溶解度对于包涵体的形成比较关键。
6. 在细菌分泌的某个阶段,蛋白质分子间的离子键、疏水键或共价键等化学作用导致了包涵体的形成。
7. 有报道表明,很多细胞包涵体具有天热酶活性。工业上经常从细菌包涵体复性,获得可溶和高活性的蛋白。
材料(MATERIALS)
r 试剂(REAGENTS)
1. 破菌液:250 mM NaCl、2.5 mM EDTA、20 mM Tris pH 8.0。
2. Wash buffer 1:250 mM NaCl、0.5% Triton X-100、2.5 mM EDTA、10 mM 2-mercaptoethanol、20 mM Tris-HCl pH 8.0。
3. Wash buffer 2:250 mM NaCl、2.5 mM EDTA、10 mM 2-mercaptoethanol、20 mM Tris-HCl pH 8.0。
4. 变性缓冲液:6 M GuHCl/8 M尿素、200 mM NaCl、10 mM 2-mercaptoethanol、2 mM EDTA、20 mM Tris-HCl pH 8.0。
5. Refolding buffer:无固定的配方,可以通过文献查询使用的复性液,最简单的复性液就是10 mM Tris。另外精氨酸,以及氧化还原体系(如氧化型谷胱甘肽:还原型谷胱甘肽等)也是经常加入的成分。本实验复性人PD-1的复性液为0.4 M L-Arg hydrochloride、2 mM EDTA、5 mM Cystamine、0.5 mM Cysteamine、0.1 M Tris pH 8.0;而PD-L1的复性液为1 M L-Arg hydrochloride、0.25 mM Oxidized Glutathione、0.25 mM Reduced Glutathione、2 mM EDTA、0.1 M Tris pH 8.0。
6. 透析液:无固定配方,可以采用20 mM NaCl、10 mM Tris或者PBS等,本实验室复性PD-1和PD-L1最终是透析到20 mM NaCl、10 mM Tris pH 8.0中。
r 实验前准备(SETUP)
冰、SDS-PAGE胶、干净的样品收集管
r 器械(EQUIPMENT)
磁力搅拌器、AKTA纯化仪、低温高速离心机、电泳槽、电泳仪
实验步骤(PROCEDURE)
1. 蛋白表达
1) 于21点左右,挑取一个生长良好的含有表达载体的表达菌克隆,接种20 mL LB/1%葡萄糖/kan培养基中,37 °C,220 rpm培养约12 h。
2) 次日9点左右,1:50接种1 L LB/kan,37 °C培养2.5 h至OD600为0.8左右,加入终浓度为0.5 mM的IPTG,37 °C诱导表达3 h。
3) 取300 μL诱导的菌液制备50 μL SDS PAGE上样缓冲液,取5 μL上样跑电泳鉴定目的蛋白是否表达。
4) 5000 g,4 °C离心10 min,收集菌体,此细菌可在-80 °C长期保存备用。
2. 破菌
1) 以1 g菌加10 mL破菌液的比例加入预冷的破菌液重悬菌体,设置超声破碎仪功率为10%,工作1秒停4秒,总工作时间10 min,冰水浴中破菌。
2) 破菌产物采用15000 g,4 °C离心30 min,弃上清。
3. 清洗包涵体
1) 采用100 mL Wash buffer 1重悬包涵体,室温磁力搅拌过夜。
2) 15000 g,4 °C离心30 min,弃上清,采用wash buffer 1重悬沉淀,室温磁力搅拌6 h。
3) 15000 g,4 °C离心30 min,弃上清,采用wash buffer 2重悬沉淀,室温磁力搅拌2 h。
4) 称量沉淀的总质量,然后采用适当体积的wash buffer 2重悬,分装至1.5 mL离心管中(保证每管含有约0.1 g沉淀),15000 g,4 °C离心20 min,去掉上清,包涵体沉淀可长期储存于-80 °C备用。
4. 包涵体变性
1) 每管包涵体加入10 mL变性缓冲液重悬,室温搅拌4 h。
2) 15000 g,4 °C离心30 min,弃沉淀,测上清蛋白浓度,4 °C存放。
5. 蛋白复性
1) 取以上10 mL蛋白变性液一滴滴加入200 mL室温下不断搅拌的复性液中(复性液中蛋白的终浓度为0.1 mg/mL左右)。
2) 放置于4 °C中孵育过夜。
3) 采用0.8 μm滤膜过滤除去沉淀,上清储存于4 °C。
4) 采用2 L透析液在4 °C下搅拌透析6 h。
5) 采用4 L透析液在4 °C下搅拌透析12 h。
6. 蛋白浓缩
1) 由于稀释和透析后蛋白溶液体积很大,浓度很低,因此有如下几种浓缩方式:
A.将蛋白溶液装在透析袋中,在透析袋外撒一些PEG20000进行浓缩,本实验室复性的PD-1和PD-L1就是这样浓缩的。
B. 采用浓缩管离心浓缩。
C. 采用离子交换柱进一步纯化并浓缩蛋白。
2) 最终将浓缩的蛋白离心除去沉淀,并测量浓度。
3) 采用24 mL分子筛进一步纯化蛋白,然后采用浓缩管浓缩蛋白,分装后-80 °C保存。
针对性建议(TROUBLESHOOTING)
1. 复性很难达到100%,因蛋白而异,能有10%的蛋白复性成功就很不错了。
2. 判断是否复性成功可以采用通过鉴定复性的蛋白的生物学功能来确定,如检测复性的PD-L1和PD-1能否相互结合。如果没有酶活检测方法,一般认为能获得较稳定单体成分,就认为复性成功。
3. 可考虑透析复性代替稀释复性,但透析前应将蛋白浓度稀释到约0.2 mg/mL,否则很可能产生大量沉淀。
4. 需要从时间、成本、体积、效率等因素,综合考虑,从而确定最优复性和下游纯化工艺。
3.2.15 SDS-PAGE凝胶电泳
简介(INTRODUCTION)
目前,蛋白质电泳分离通常采用聚丙烯酰胺凝胶作为电泳介质。蛋白质在电场力的牵引下,在凝胶形成的孔径内进行迁移。凝胶浓度越高,孔径越小。凝胶孔径的大小与蛋白质大小、电荷以及形状相结合,最终决定了蛋白质的迁移速度。
根据凝胶中是否加入蛋白变性剂,蛋白电泳可分为变性电泳和非变性电泳。变性电泳(常添加有0.1%的SDS)可用来估计多肽或蛋白质的纯度和分子量,而非变性电泳(常添加有2-巯基乙醇或二硫苏糖醇等还原剂)则可用来确定蛋白质的亚基数目和大小。非变性电泳可用来检测和分离“天然”状态的蛋白。
材料(MATERIALS)
r 试剂(REAGENTS)
1 M Tris-HCl(pH 6.8)、30% Acrylamide、10% SDS、10% APS、TEMED、超纯水、SDS-loading buffer、考马斯亮蓝G250染液等
r 器械(EQUIPMENT)
梯度混合器、电泳仪、电源、电泳槽等
实验步骤(PROCEDURE)
► 配制梯度变性胶 ►预计时间消耗:4小时
梯度胶由两种组分混合而成,轻密度部分(light)和重密度部分(heavy)。我们一般使用的是4~15% 梯度胶,因此轻密度部分是4%,重密度部分是15%。也就是用混匀器时,重密度部分会缓缓加入到轻密度的腔室中,胶由下而上灌制,因此胶的上层密度比下层密度小。
SDS-PAGE梯度胶的配方如下:
| Heavy(for 65 mL) | Light(for 65 mL) | ||||
Concentration | 20% | 18% | 15% | 12% | 4% |
|
ddH2O (mL) | 3.0 | 7.5 | 23 | 20.5 | 37 |
|
1 M Tris-Hcl pH 6.8 (mL) | 17.5 | 17.5 | 19 | 17.5 | 19 |
|
30% Acrylamide (mL) | 43.3 | 39 | 29 | 26 | 15 |
|
10% SDS (mL) | 0.7 | 0.7 | 0.7 | 0.7 | 0.77 |
|
10% APS(fresh)(mL) | 0.7 | 0.7 | 0.7 | 0.7 | 0.77 |
|
TEMED (μL) | 30 | 30 | 25 | 30 | 0.025 |
|
各种buffer的储液配制方法:
30% acrylamide(for 1 L):292 g acrylamide、8 g bis-acrylamide,4 °C避光保存。
10% APS(w/v):配制50 mL时,需加入5 g APS,加入50 mL蒸馏水,溶解后4 °C避光保存。
10% SDS(w/v):配制100 mL时,需加入10 g SDS,加入100 mL蒸馏水,溶解后常温保存即可。
▲ 关键步骤:
♫ 梯度胶由专人负责配置,故具体制胶过程略去不表,仅述制胶中需要注意的一些要点:
♫ 胶板填装时,要使用塑料胶片填充严实。检查是否漏液和排除管道内空气时,要注意连接处是否紧密;
♫ 制胶需要调节TEMED的加入量,以调节凝胶凝结的速度,添加量视气温而定。温度越高凝固越快。
♫ 灌胶过程中,需要转子以适当转速在梯度混合器内的低浓度胶中进行转动,以充分混匀来自梯度混合器另一侧的高浓度胶。同时,为了达到充分混匀的目标,将梯度混合器的黑色混合开关全部打开,而凝胶流通管道上的开关是由配胶者控制总体速度。玻璃板的液面不能上升太快,尽量缓慢流出。这对凝胶在水平方向上的浓度均一性尤为关键,直接影响到制胶的成败。
♫ 当梯度混合器中的液体胶快流完时,提前关闭开关,剩余的弃之。因为往往最后会残留很多气泡,不能让气泡进去。
跑变性胶 ►预计时间消耗:1小时
1. 根据计划跑胶的样品数量计算所需要的胶孔数目,选择合适样品孔的凝胶(节约原则),装在电泳仪中。胶板由一长一短两块儿板组成,短的那块朝里侧放置。
2. 将running buffer倒在电泳槽中,液面恰好在两种高度的胶板之间。
3. 上样:提前煮好的蛋白样品,用白色的小枪头缓缓加入凝胶上样孔中。样品加入体积一般在5~7 μL左右,如果样品较浓时3 μL足矣。如果上样量太大,会降低电泳分辨率。
4. 电泳:盖上电泳仪上盖,150 V电压运行48 min(如果着急看结果,可以200 V电压电泳24~30 min)。
▲关键步骤:
♫ 注意loading buffer中的溴酚蓝示综染料不要跑尽,即不要将溴酚蓝进到running buffer中,因为我们的buffer是要回收利用的。
♫ 染色:将胶板取出,用自来水将表面的气泡冲掉,用拆胶板把玻璃板撬开,将含有胶的玻璃板直接放在盛有自来水的染盒中,凝胶顺水冲落至染盒。
♫ 将染盒至于微波炉内,高火将水煮沸2分钟。此步骤的目的是将胶上的SDS煮掉,因为SDS可以与考马斯亮蓝竞争性地与丙烯酰胺结合。重复1次,尽量把SDS洗干净。
♫ 加入G250染液,在微波炉高火煮3分钟,将染液倒至染液回收缸中。用自来水将胶上的浮色洗净,加自来水煮沸至少3次,直到背景很浅为止。
♫ 拍照:使用白光投射或者白光反射,根据实际情况调节光圈与曝光时间。一般光圈用3,曝光时间在30+ ms左右。照胶仪拍摄的照片是黑白的,因此也可以用自己的手机进行拍照,效果更好。
♫ 清理:煮胶间隙,把所用的凝胶系统清洗干净并放置于本实验室规定放置的地方,清理实验台面。
针对性建议(TROUBLESHOOTING)
如果需要检测的蛋白很大(超过100 kDa)时,如做crosslinking实验时往往会遇到这种情况,需要用4~12%梯度胶,尽量让大分子量的组分分开。样品浓度过高时,建议稀释样品制样。一定注意电极的正确插入。
3.2.16 GraphPad Prism软件的使用
简介(INTRODUCTION)
GraphPad Prism是一款集数据分析和作图于一体的数据处理软件,它可以直接输入原始数据,自动进行基本的统计分析,同时产生高质量的科学图表。GraphPad Prism适用于Windows和Mac平台,现在最新的版本是GraphPad Prism7,GraphPad Prism已经被广泛的被各类生物学家,以及社会和物理学家所使用,也被广泛的被本科生和研究生所使用。
► 软件要求(SOFTWARE REQUIREMENT)
Windows或者Mac操作系统
GraphPad Prism软件(down from www.graphpad.com)
基本使用步骤(PROCEDURE)
这里以实验室常用的蛋白纯化图为例讲解GraphPad Prism软件的使用。
 | |||
| |||

1.新建表格。打开Prism5软件,点击choose a graph为散点图,然后点击Enter and plot a single Y value for each point,最后点击Create。
|

2. 输入数据。根据AKTA输出的有关第一次镍柱洗脱的Excel的数据,复制其中A280的横纵坐标数据,粘贴到Prism5生成的表格中,点击data查看生成的图形。

3. 点击文档操作模块中的New按钮,新建一个新的New data table and graph表格(根据第一步的操作),复制其中Elution Buffer的梯度数据,粘贴到新生成的表格中,点击Data查看生成的图形。

4.两个图形的叠加。双击第一个生成的图形,选择Data Sets on Graph,然后点击Add,出现右侧的对话框,点击Right,最终点击OK叠加两个图形。

5.修改调整图片。双击生成的曲线,出现下图的对话框,点击Show connecting line/curve,下一步调整曲线的颜色和粗细,点击OK。一般选择A280为蓝色,Elution Buffer 梯度线为红色。
6.调整横纵坐标。左边纵坐标为A280(mAu)右边纵坐标为B%,横坐标为Elution Volume(mL),最后是图注为纯化日期+蛋白名称。(若需要修改图形的某一部分,双击即可启动该部分的编辑模式)
针对性建议(TROUBLESHOOTING)
|

1. 如果AKTA输出的Excel数据有负数,最好在Excel中加入某个合适的数值以至于纵坐标数据都为正数,没有负数,让纯化图更加美观。
2. 在文档的操作模块中,可以将制作好的蛋白纯化图格式保存为一个模板,当下次使用相同的格式时,可以在依据下图指示复制到你的新图上。
3. 此外,GraphPad Prism还具有绘制热图、生存曲线、柱状图、箱式图、散点图、趋势图、计算拟合曲线和IC50等功能。
3.3 筛选与蛋白互作的分子
筛选与蛋白质相互作用的实验技术有多种,这里列举几项本实验室的现有技术。
3.3.1 酵母双杂交技术从文库中筛选与蛋白X结合的蛋白
简介(INTRODUCTION)
我们的酵母双杂交体系是依赖酵母转录因子GAL4的系统。GAL4包括两个彼此分离的但功能必需的结构域:位于N 端1-147位氨基酸残基区段的DNA结合域(DNA binding domain,DNA-BD)和位于C端的768-881位氨基酸残基区段的转录激活域(Activation domain,AD)。DNA-BD能够识别位于GAL4效应基因GAL4- responsive gene的上游激活序列(Upstream activating sequence,UAS),并与之结合。而 AD则是通过与转录机器(transcription machinery)中的其他成分之间的结合作用,以启动UAS下游的基因进行转录。DNA-BD和AD单独作用并不能激活转录反应,只有当二者在空间上充分接近时,才呈现完整的GAL4转录因子活性并可激活UAS下游启动子,使启动子下游基因得到转录。见下图:
|
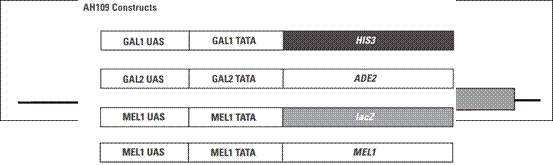
我们实验室用的酵母菌菌株是AH109,它含有4个报告基因,如下图:
HIS3报告基因启动后,酵母可以在组氨酸缺失的平板上生长;ADE2报告基因启动后,酵母可以在腺嘌呤缺失的平板上生长;lacZ和MEL1报告基因是平行的,两个基因上游都是由MEL1 UAS和MEL1 TATA区域同时控制的,区别在于lacZ的产物是β-半乳糖苷酶,其底物是X-gal;而MEL1的产物是a-半乳糖苷酶,其底物是X-a-gal。
材料(MATERIALS)
r 试剂(REAGENTS)
1. YPDA培养基
YPDA培养基(1 L) |
|
蛋白胨 | 20 g |
酵母粉 | 10 g |
0.2%硫酸腺嘌呤 | 15 mL |
琼脂粉(固体) | 20 g |
40%的葡萄糖溶液 | 50 mL |
2. SD/-Trp培养基
SD/-Trp培养基(1 L) |
|
酵母氮源(含硫酸铵) | 6.7 g |
10×-Trp DO Supplement | 100 mL |
琼脂粉(固体) | 20 g |
40%的葡萄糖溶液 | 50 mL |
▲ 注:100 mL的10×-Trp DO Supplement中含有-Trp DO Supplement量为0.74 g。葡萄糖溶液单独配制和灭菌(121 °C,15 min),除葡萄糖以外的药品加入去离子水950 mL,调节pH至6.5,121 °C高压灭菌15 min,冷却至55 °C左右,再加入灭菌的40%的葡萄糖溶液50 mL(葡萄糖最终含量为2%)。
3. SD/-Trp/-Lue培养基(1 L)
药品名称 | 药品用量 |
酵母氮源(含硫酸铵) 10×-Trp/-Lue DO Supplement 琼脂粉(固体) 40%的葡萄糖溶液 | 6.7 g 100 mL 20 g 50 mL |
注:100 mL的10×-Trp/-Lue DO Supplement中含有-Trp/-Lue DO Supplement量为0.64 g。
4. SD/-Trp/-Lue/-His/-Ade培养基(1 L)
药品名称 | 药品用量 |
酵母氮源(含硫酸铵) 10×-Trp/-Lue/-His/-Ade DO Supplement 琼脂粉(固体) 40%的葡萄糖溶液 | 6.7 g 100 mL 20 g 50 mL |
注:100 mL的10×-Trp/-Lue/-His/-Ade DO Supplement中含有-Trp/-Lue/-His/-Ade DO Supplement 0.6 g。
5. SD/-Trp/-Lue/-His/-Ade/X-α-Gal培养基(1 L)
药品名称 | 药品用量 |
酵母氮源(含硫酸铵) | 6.7 g |
10×-Trp/-Lue/-His/-Ade/X-α-Gal DO Supplement | 100 mL |
琼脂粉(固体) 40%的葡萄糖溶液 20 mg/mL X-α-Gal | 20 g 50 mL 2 mL |
▲ 注:20 mg/mL X-α-Gal是用二甲基甲酰胺(DMF)溶制的。除葡萄糖和X-α-Gal以外的药品加入去离子水948 mL,调节pH至6.5,121 °C高压灭菌15 min,降温后再加入40%的葡萄糖溶液50 mL和20 mg/mL X-α-Gal 2 mL。
6. 10×TE buffer:0.1 M Tris-HCl, 10 mM EDTA pH 7.5. Autoclave.
7. 10×LiAc:1 M lithium acetate (Sigma Cat No. L-6883) Adjust to pH 7.5 with diluted acetic acid and autoclave.
8. 50%PEG3350:prepare with sterile deionized H2O; if necessary, warm solution to 50°C to dissolve PEG.
9. 1.1×TE/LiAc Solution:Prepare fresh just prior to transformation using the stock solutions provided. Combine 1.1 mL of 10×TE Buffer with 1.1 mL of 1 M LiAc (10×). Bring the total volume to 10 mL using sterile, deionized H2O.
10. PEG/LiAc Solution:
| Final Conc. | To prepare 10 mL of solution |
PEG 3350 | 40% | 8 mL of 50% PEG 3350 |
TE buffer | 1× | 1 mL of 10×TE buffer |
LiAc | 1× | 1 mL of 1 M LiAc (10×) |
11.0.9% (w/v) NaCl Solution:Dissolve 0.9 g of NaCl in 100 mL of deionized H2O and filter-sterilize the solution.
r 实验前准备(SETUP)
冰,灭菌的50 mL、15 mL及1.5 mL离心管,所有试剂及培养基配制及灭菌
r 器械(EQUIPMENT)
Thermo 离心机、金属煮样器、台式离心机、30 °C摇床、30 °C培养箱、超净台等。
实验步骤(PROCEDURE)
1.筛选前准备
1) 构建诱饵质粒:将诱饵克隆到V104/pGBKT7质粒中去。
2) 扩增AD Fusion Library。
3) 检测自激活,总的来说就是分别将诱饵Small-Scal单独转化AH109,在SD/–Trp/X-α-Gal和SD/–Leu/X-α-Gal板上铺板观察是否变蓝。以下是具体操作步骤:
A.制备AH109感受态细胞
a) 复苏保种的AH109,30 °C倒置培养3~5天,从YPDA平板上挑取酵母单克隆(直径2~3 mm,<4周)接种于3 mL YPDA液体培养基,30 °C,250 rpm摇床培养8~12 h。
b) 转5 μL过夜培养的酵母菌液于50 mL YPDA液体培养中继续相同条件摇床培养16~20 h,直至OD600达到0.15~0.3。
c) 20 °C,1500 g离心5 min收集菌体,弃上清,用100 mL新鲜的YDPA液体培养基重悬菌块,30 °C继续培养3~5 h,直到OD600达到0.4~0.5。
d) 将菌液分装于两个50 mL无菌离心管中,室温1500 g离心5 min收集菌体,弃上清后分别用30 mL灭菌的ddH2O重悬,清洗一次。
e) 将菌液分装于两个50 mL无菌离心管中,室温1500 g离心5 min收集菌体,弃上清后分别加入1.5 mL 1.1×TE/LiAc重悬菌体,将菌液分别转移至两个1.5 mL的无菌EP管中,室温5000 rpm离心30 s。
f) 弃上清后分别用0.6 mL的1.1×TE/LiAc重悬菌块,即为酵母感受态细胞,最好立即使用,或置于冰面1 h内使用。
B. 诱饵质粒小规模转化酵母感受态
a) 先向1.5 mL无菌EP管中加入100 ng诱饵质粒,再加入5 μL灭活的Carrier ssDNA轻轻混匀,随后加入50 μL感受态细胞轻混后在加入500 μL PEG/LiAc,30 °C孵育30 min,每隔10 min轻轻弹动EP管使菌液混匀。
b) 孵育30 min后加入20 μL DMSO并混匀,42 °C水浴热激15 min。每隔5 min,轻轻涡旋是菌液均匀。
c) 5000 rpm离心30 s收集酵母,弃上清后加入1 mL YPD重悬,30 °C摇床培养30 min。
d) 5000 rpm离心30 s收集酵母,用1 mL 0.9% (w/v) NaCl重悬,取10 μL和1 μL重悬为100 μL铺SD/-Trp铺板计算转化效率。
e) 从SD/-Trp板上挑取转化入诱饵质粒的酵母AH109单克隆,分别划线于SD/–Trp/X-α-Gal、SD/–Trp/-His、 SD/–Trp/-Ade培养基上,30 °C倒置培养3~5天,检测MEL1、HIS3、ADE2报告基因的表达。
4) 验证转化的酵母质粒:
A. 通过PCR验证,具体是抽提酵母质粒(方法参照OMEGA酵母质粒抽提试剂盒说明书),利用对应抗原cDNA序列的特异性引物PCR扩增抗原序列,从而验证转进酵母的质粒是否抗原质粒。
B. 通过WB验证,具体是分别将诱饵和猎物质粒单独转化AH109,Western blotting验证融合蛋白的表达,一抗分别为抗c-Myc和HA epitope tags的抗体和抗AD的抗体,采用未转化组为对照。
C. 检测3-AT使用的浓度:
转化诱饵质粒进入AH109后,再采用Library-scale转化文库质粒进入进行酵母双杂交筛选,涂布在含有0、2.5、5、7.5、10、12.5、15、20 mM 3-AT的SD/-Trp/-Leu/-His培养基上涂布生长,观察能够抑制背景生长,且阳性克隆数量和大小适中的浓度作为使用浓度进行筛选。如不想摸索,可直接使用10 mM 3-AT开始进行实验,根据克隆数和大小增加或减少2.5 mM。
2. 筛选
制备足够数量的感受态细胞,按照Yeastmaker™ Yeast Transformation System 2 User Manual操作,1管感受态为600 μL,可同时制备多管,如10管,即6 mL,以下是制备和转化6 mL感受态的具体步骤:
1)制备酵母感受态
A. 从SD/-Trp平板上挑取酵母单克隆(直径2~3 mm,<4周)接种于5 mL SD/-Trp液体培养基,30 °C,220 rpm摇床过夜培养。
B. 测量过夜培养酵母菌液OD600(需要稀释5倍),转适当体积(2~10 mL)过夜培养的酵母菌液于250 mL SD/-Trp液体培养中(500 mL三角烧瓶),起始OD600约0.1,继续相同条件摇床培养4~6 h,直至OD600达到0.4~0.5。
C. 20 °C 1500 g离心5 min收集菌体,弃上清后用300 mL灭菌的ddH2O重悬团块,清洗一次。
D.将菌液分装于200 mL无菌离心瓶中,室温5000 g离心5 min收集菌体,弃上清后用15 mL 1.1×TE/LiAc重悬菌体。
E.将菌液转移至50 mL无菌离心瓶中,室温1500 g离心5 min收集菌体,弃上清后用6 mL 1.1×TE/LiAc重悬菌体,即为酵母感受态细胞,最好立即使用,或置于冰面1 h内使用。
2) 文库质粒Library-Scale转化含有诱饵质粒的酵母感受态细胞
A.向1.5 mL无菌EP管中加入60 μg质粒,再加入200 μL heat denatured salmon sperm DNA (ssDNA) (5 mg/mL,先加热至95 °C,然后放冰上待用) 轻轻混匀,将以上混合的DNA加入上面制备的6 mL的感受态酵母中,混匀后加入25 mL PEG/LiAc,利用15 mL移液管混匀,再颠倒混匀5次,30 °C水浴孵育45 min,每隔15 min颠倒混匀一次。
B. 孵育30 min后加入1.6 mL DMSO并颠倒混匀,分装至3根15 mL离心管,42 °C水浴热激20 min。每隔10 min颠倒混匀一次。
C.20 °C 2000 g离心5 min收集酵母,弃上清后加入30 mL YPD重悬,转移至灭菌的200 mL三角烧瓶,30 °C,220 rpm培养90 min。
D. 转移至灭菌的50 mL离心管,1500 g离心5 min,弃上清,用10 mL 0.9% (w/v) NaCl重悬,取10 μL分别稀释10-2和10-3后取10 μL重悬为100 μL铺SD/-Trp/-Leu板计算转化效率。
E. 分别取500 μL平铺于含有SD/-Trp/-Leu/-His/10 mM 3-AT大培养皿(150 mm)上,置30 °C培养箱,连续培养7天后观察生长的阳性克隆。
F. 将阳性克隆划线于SD/–Leu/–Trp/X-α-Gal上,培养4到6天后,再挑取一个蓝色克隆划线SD/–Leu/–Trp/X-α-Gal上,培养4到6天后挑取一个蓝色克隆在SD/-Trp/-Leu/-Ade/-His/10 mM 3-AT/x-α-gal上划线,观察是否生长和变蓝。将变蓝的克隆转移至画着格子的SD/-Trp/-Leu/-Ade/-His/10 mM 3-AT/x-α-gal上进一步鉴定,保鲜膜封口,4 °C保存,可保存4周。
3. 假阳性鉴定:
1) 抽提酵母质粒,转化大肠杆菌,Ampicillin抗性筛选即可得到含文库scFv质粒的克隆,培养含有阳性文库质粒的单克隆菌并抽提质粒(OMEGA大肠杆菌质粒抽提试剂盒)。送样测序,排除重复。
2) 分别制备含有pGBKT7、pGBKT7-laminC、pGBKT7-抗原的AH109感受态细胞,Small-scale将待确定阳性的克隆猎物质粒分别转化入含有pGBKT7、pGBKT7-laminC、pGBKT7-诱饵的AH109中,涂布于SD/–Leu/–Trp /–Ade/–His /X-α-Gal/10 mM 3-AT培养基上培养7天后观察生长和变蓝情况,真阳性scFv质粒只有转化进入含有pGBKT7-抗原的AH109中才能在SD/–Leu/–Trp /–Ade/–His/X-α-Gal/10 mM 3-AT培养基上生长和变蓝,若转化进入含有pGBKT7或者pGBKT7-laminC的AH109也能在SD/–Ade/–His/–Leu/–Trp/X-α-Gal/10 mM 3-AT培养基上生长和变蓝,则定义为假阳性。
针对性建议(TROUBLESHOOTING)
1. AH109菌株在普通YPD平板上生长缓慢,出现略带粉色克隆。最好用YPDA板,克隆为白色隆起,且生长较快。
2. 此方法采用添加了3-AT的SD/-Leu/-Ade/-His平板进行筛选,所得的阳性克隆均是相互作用很强的克隆。如果对阳性要求不强可改用不加3-AT的SD/-Leu/-Trp/-Ade/-His平板筛选,得到的克隆数将很多。
3. 培养酵母的平板均无抗性,一定要注意不要染菌。
4. 酵母培养时间较长,可以使用保鲜袋将平板密封后培养,以免水分损失太多。
3.3.2 噬菌体展示
 简介(INTRODUCTION)
简介(INTRODUCTION)
噬菌体展示技术的基本原理是:将编码外源多肽的DNA片段与噬菌体衣壳蛋白的编码基因融合,从而将外源基因表达并展示在噬菌体的表面(如下图所示),被展示的多肽或蛋白可保持相对的空间结构和生物活性。用一个靶标蛋白质或其它分子去筛选噬菌体展示库,从而分离出库里的某些能结合靶标的噬菌体,进而获得能与结合靶标的蛋白或者多肽。噬菌体展示已广泛用于多肽类药物的研发当中,目前已经有超过20个多肽及抗体药物是采用该技术筛选获得的。
材料(MATERIALS)
r 试剂(REAGENTS)
1. PBS缓冲液(1 L,pH 7.4)
药品名称 | 药品用量(g) |
Na2HPO4·12H2O | 2.9 |
NaCl | 8 |
KCl | 0.2 |
KH2PO4 | 0.2 |
121 °C灭菌20 min,4 °C储存。
2.TYE固体培养基(1 L)
药品名称 | 药品用量(g) |
蛋白胨 | 10 |
酵母粉 | 5 |
NaCl | 8 |
琼脂粉 | 15 |
使用1 L ddH2O溶解,121 °C灭菌20 min,待冷却至50 °C左右时后倒平板,即获得无抗生素TYE平板。使用800 mL ddH2O溶解,121 °C灭菌20 min,待冷却至50 °C左右时加入200 mL 20%(m/V)葡萄糖(G)溶液和抗生素,混匀后倒平板,即获得含有抗生素和4%G的TYE平板。
3. 2×TY培养基(1 L)
药品名称 | 药品用量(g) |
蛋白胨 | 16 |
酵母粉 | 10 |
NaCl | 5 |
121 °C灭菌20 min,4 °C储存。
4. 胰酶溶液:使用TBSC缓冲液将胰酶粉末溶解,配制为10 mg/mL的储存液,-20 °C储存。使用前使用TBSC缓冲液稀释为0.5 mg/mL工作浓度。
5.20% PEG/NaCl溶液(500 mL)
药品名称 | 药品用量(g) |
PEG6000 | 100 |
NaCl | 73 |
121 °C灭菌20 min,4 °C储存。
6.TBSC缓冲液(1 L,pH 7.4)
药品名称 | 药品用量(g) |
Tris Base | 1.5 |
NaCl | 8 |
CaCl2 | 0.15 |
121 °C灭菌20 min,4 °C储存。
7. OMEGA(广州飞扬生物公司)M13 isolation kit
8. SOC培养基:2% (W/V) Tryptone、0.5% (W/V) Yeast Extract、0.05% (W/V) NaCl、2.5 mM KCl 10 mM MgCl2、20 mM glucose。121 °C灭菌20 min,分装50 mL/管,放在-20 °C冰箱中保存。
9. Gel filtration buffer(GFB):250 mM NaCl、20 mM Tris pH 8.0,121 °C高温灭菌。
r 实验前准备(SETUP)
所有试剂及培养基配制及灭菌,免疫管,96孔免疫板等
r 器械(EQUIPMENT)
离心机、全温度摇床、培养箱、超净台等
实验步骤(PROCEDURE)
1. KM13 Helper phage扩增
1) 从新鲜划线的无抗生素TYE平板上挑取一个TG1单克隆接种到5 mL 2x TY培养基中,37 °C、220 rpm震荡培养12 h。
2) 取50 μL菌液重新接种到5 mL 2x TY培养基中,37 °C、220 rpm培养约1.5 h至OD600为约0.5。
3) 使用2×TY稀释Helper phage 至4 × 1010 pfu/mL,取10 μL稀释液加入200 μL以上制备的TG1菌液中,轻轻混匀, 37 °C水浴1 h,让Helper phage侵染进入TG1。
4) 将上述TG1菌液加入到3 mL 42 °C水浴的H-Top琼脂培养基中混匀,铺在37 °C预热的TYE平板中,室温冷却凝固后,37 °C培养约12 h。
5) 挑取1个TYE平板上的空斑接种5 mL 新鲜生长的TG1菌液中(OD600约0.5),37 °C、220 rpm震荡培养2 h。
6) 将此5 mL菌液转移至盛有500 mL 2×TY培养基的2 L三角烧瓶中,37 °C、220 rpm震荡培养1 h,然后加入终浓度为50 μg/mL的kanamycin,30 °C、250 rpm培养过夜。
7) 4 °C,16000 g离心15 min,取400 mL上清至500 mL烧杯中,加入100 mL 20% PEG/NaCl,混匀后冰浴1 h,让噬菌体析出。
8) 4 °C,16000 g离心15 min,弃上清,沉淀加入8 mL PBS溶解,转移至15 mL 离心管中,加入2 mL 20% PEG/NaCl,混匀后置于冰浴中20 min,让噬菌体析出。
9) 4 °C,16000 g离心15 min,弃上清,沉淀加入5 mL PBS溶解,16000 g、4 °C离心15 min,上清转移至一个新的15 mL离心管中,加入终浓度为15%的甘油,
10) 用0.45 μm滤膜过滤,分装后-80 °C保存。
11) 滴度测量:
A. 取1 μL扩增的helper phage溶液加入1 mL PBS中混匀(1:1000稀释),从1:1000稀释液中取10 μL加入990 μL PBS中混匀(1:100稀释),再依次按照此法(1:100)稀释4次,共稀释了5次。
B. 分别从以上第4和5次的稀释液中取50 μL加入1 mL TG1(OD600约0.5),混匀,37 °C水浴30 min。
C. 分别加入到3 mL 42 °C H-Top琼脂培养基中混匀,铺在37 °C预热的TYE平板中,室温冷却凝固后,37 °C培养约12 h。
D. 分别数平板上的空斑数,并按照以下公式计算滴度
滴度(pfu/mL) =  x
x 
2.制备噬菌体展示文库
下面以制备VHH噬菌体展示文库为例,其中pR2为噬菌体质粒。
1)制备dU-ssDNA
A. 将pR2-VHH转化E. coli CJ236感受态,涂布在LB/Amp平板上,37 °C培养过夜。
B. 挑取一颗生长良好的单克隆接种于2 mL 2×TY/ cmp/Amp中,加入2×1010 pfu的KM13,37 °C、200 rpm培养2.5 h。
C. 加入Kan,37 °C、200 rpm培养5 h。
D. 5000 g离心5 min,沉淀转移至60 mL 2×TY/cmp/Amp/Kana/uridine培养基中。37 °C、200 rpm培养5~10 h(至OD600为0.6~0.8之间)。
E. 分装至2个50 mL离心管中,4800 g、4 °C离心15 min,上清转移至新的离心管。共加入15 mL PEG/NaCl,冰浴1 h。
F.17000 g、4 °C离心15 min,弃上清。沉淀用6 mL PBS重悬,分装至4个2 mL离心管中,10000 g、4 °C离心5 min,上清转移至新的2 mL离心管并重复离心一次。
G. 转移上清至装有1.5 mL PEG/NaCl的15 mL离心管中,混匀,冰浴10 min。
H. 用真空抽滤的方法将以上孵育液全部滤过2个HiBindR M-13 column,随后10000 rpm离心30 s,弃滤液。
I. 加入500 μL MPB buffer至柱子中,室温孵育1 min ,10000 rpm离心30 s,弃滤液。
J. 重复上步。
K. 加入700 μL SPW Wash Buffer(使用前加入乙醇),10000 rpm离心30 s,弃滤液。
L. 重复上步。
M. 15000 rpm离心2 min使柱子干燥。
N. 将柱子转移至1.5 mL的离心管,加入50 μL 超纯水(pH在7.5~8.0之间)至柱子中央,室温孵育2 min,15000 rpm离心1 min。
O.测量dU-ssDNA浓度,跑琼脂糖胶验证(设置双链pR2-VHH作为对照)。
2)合成文库CCC-dsDNA
A. 设计并合成简并寡核苷酸,目前本实验是在VHH的CDRs区引入NNK随机化,简并寡核苷酸5’端和3’端均有20的碱基与模板匹配,其中CDR1和2均只合成1条简并引物,而CDR3合成3条简并引物以引入不同的长度的CDR3。引物在生工合成。
B.寡核苷酸磷酸化:在PCR管中混合3.9 μL 10uM的CDR1/2寡核苷酸、2.0 μL 10×TM buffer、2.0 μL 10 mM ATP、1.0 μL 100 mM DTT,20 U T4 polynucleotide kinase (2 μL),加入水补齐到20 μL,不同的寡核苷酸需要单独进行。在PCR管中混合1.56 μL 10uM的CDR3-1\2\3寡核苷酸、0.8 μL 10× TM buffer、0.8 μL 10 mM ATP、0.4 μL 100 mM DTT,8 U T4 polynucleotide kinase (0.8 μL),加入水补齐到8 μL,不同的寡核苷酸需要单独进行。37 °C孵育1 h,立即用于下一步。
C.取20 μg dU-ssDNA(约13 pmol),25 μL 10×TM buffer,磷酸化的寡核苷酸CDR1\2各20 μL,CDR3-1\2\3各6.67 μL,加入水至250 μL。注意:此中,寡核苷酸:dU-ssDNA的摩尔比为3:1。分装到5个PCR管中。
D.90 °C孵育3 min,50 °C孵育3 min,20 °C孵育5 min。此时寡核苷酸已结合到dU-ssDNA上(即退火)。将5管PCR管中的溶液转移至1个1.5 mL离心管中。
E.加入10 μL 10 mM的ATP,25 μL 40 mM的dNTP,15 μL 100 mM DTT,5 μL T4 DNA连接酶(400 U/μL),3 μL T7 DNA polymerase(10 U/μL)。混匀,20 °C孵育过夜。
F. 采用3个qiagen PCR回收试剂盒中的DNA结合柱子回收反应产物,分别采用40 μL ddH2O洗脱。
G. 跑琼脂糖胶分析:取0.5 μg以上洗脱的DNA,以dsDNA及dU-ssDNA作为对照,跑琼脂糖胶电泳,成功合成的CCC-dsDNA呈现3条带,如下图所示。
 |
H. 测量洗脱的CCC-dsDNA浓度(A260 = 1.0 for 50 ng/μL of dsDNA)。回收的总量必须大于20 μg,回收的DNA可以用于下步,或-20 °C长期储存。
3) 制备E. coli TG1电转化感受态
A. 挑取一个新鲜的TG1克隆接种于3 mL 2×TY,37 °C、220 rpm培养6 h。
B. 1:100重新接种250 mL 2×TY,37 °C,200 rpm培养1 h 40 min至OD600为约0.6~0.8。
C. 接下来的步骤均于冰面操作,所有的溶剂和仪器均需要提前预冷。
D. 采用离心瓶,3500 g,4 °C离心15 min。弃上清,菌体用250 mL 10%的甘油重悬。
E. 采用离心瓶,3500 g,4 °C离心15 min。弃上清,菌体用250 mL 10%的甘油重悬,摇晃40次后置离心机中5 min,再次摇晃40次。
F. 采用离心瓶,3500 g,4 °C离心15 min。弃上清,菌体用100 mL 10%的甘油重悬。
G. 采用离心管,3500 g,4 °C离心15 min。彻底弃上清。菌体用1 mL 10%的甘油,用移液器轻轻吹打重悬。
H. 分装为100 μL每管(约12管),储存于-80 °C。
4) CCC-dsDNA转化电转化TG1
A. 将10个0.1 cm gap电击杯放置超净台吹干后于冰面预冷,将CCC-dsDNA分装0.25 μg/管,合计10管放置冰面,取10管感受态(100 μL)置于冰面解冻。
B. 取感受态100 μL加入1管装有CCC-dsDNA的管中,轻轻吹打混匀,不要产生气泡,合计10管。
C. 将混合液分别转移至10个预冷的电击杯中,设置电转仪2.5 kV field strength,6 ms进行电击。
D. 电击后立即加入1 mL 37 °C预热的SOC培养基,然后转移至10 mL离心管,电击杯分别用1 mL SOC冲洗2次,冲洗液一并加入试管中(合计3 mL)。合计操作10次。
E. 37 °C、220 rpm培养45 min。
F. 将所有10管菌液混匀后按如下方式稀释,从3~7管中分别取100 μL涂布TYE/Amp平板,以测量转化效率及获得转化子的数量。

G.将所有菌液平铺于100块150 mm的TYE/4%葡萄糖/Amp平板中,37 °C培养12 h。
H. 计算转化效率,并确定20 μg CCC-dsDNA转化能获得的转化子数量,即文库大小。
I. 每个150 mm平板加入5 mL 2×TY,用涂布棒将菌落刮下来,混匀后加入100%甘油至终浓度为20%。分装1.5 mL每管至2 mL离心管中,液氮速冻后储存于-80 °C冰箱。这就是文库菌液。
5) 文库phage制备
A. 取出一管冻存的文库菌液于冰面解冻,取适量体积(约0.5-1 mL)加入250 mL 2×TY/4%葡萄糖/Amp中,测量OD600应约为0.1。37 °C,220 rpm培养1.5~2 h至OD600为0.5,加入1012 pfu的KM13辅助噬菌体,37 °C水浴1 h。
B. 4500 g离心10 min,弃上清,沉淀用2×TY/0.1%葡萄糖/Amp/Kan重悬,25 °C、220 rpm培养16 h。
C.4500 g离心30 min,取400 mL上清加入100 mL 20% PEG/NaCl,混匀后冰浴1 h。
D. 4500 g、4 °C离心30 min,弃上清,沉淀加入20 mL PBS涡旋重悬。
E. 分装至2 mL离心管中,15000 g高速离心5 min,再转移上清至一根新的 50 mL离心管中。
F. 加入5 mL 100%甘油并混匀,用0.45 μm针头滤器过滤,测量OD260值并按如下公式计算滴度:pfu/mL = OD260 × 100 × 22.14 × 1010。
G. 分装200 μL/管,储存于-80 °C冰箱。
3. 从文库中富集阳性噬菌体
1) 第1轮筛选
A. 包被抗原
a) 取-80 °C保存的抗原于室温水中快速解冻。
b) 取0.4 mg用含有1 mM EDTA 的Gel filtration buffer(GFBE)稀释至4 mL,加入一根免疫管中,用石蜡膜封口。另设一管不加抗原的对照管,加入GFBE。
c) 放置4 °C过夜(16 h)包被。
B.封闭
a) 新鲜配制适当体积的5%脱脂牛奶(用GFBE溶解)。
b) 用5 mL移液器倾斜约30度角向各免疫管中加入5 mL GFBE,倒掉后倒置于纸巾上约5~10 s,然后再在纸巾上敲打10~20次以除去免疫管中的溶液,此为wash 1次。合计wash 2次。
c) 加满 5%脱脂牛奶,用石蜡膜封口。
d) 室温(25 °C)放置3 h封闭。
C. 孵育抗体phage
a) 取1根空白免疫管,加入4 mL新鲜配制的5%脱脂牛奶。加入171027制备的纳米抗体文库phage,input量为5 ×1012 pfu(150 μL),石蜡膜封口,室温颠倒混匀孵育30 min~1 h。
b) 对照管用GFB洗2次,加入上面的孵育液,室温颠倒孵育1 h。
c) 包被抗原管用GFB清洗2次,加入上面的孵育液,室温颠倒孵育1 h。
d) 依次对对照管和抗原管进行washing,采用GFBT(含0.1% Tween 20)各清洗10次。
D. Elution
a) 取1管Trypsin (10 mg/mL, 100 μL)加入PBS稀释,一般是100 μL母液加1900 μL PBS,混匀。
b) 分别取500 μL每管加入免疫管中,颠倒混匀15 min后转动管子45度再次消化15 min,合计消化1 h。
E.测量滴度和扩增
a) 接种一个TG1单克隆至2 mL 2TY中,37 °C、220 rpm培养8 h。
b) 1:100重新接种5 mL 2TY中,37 °C、220 rpm培养1.5 h。
c) 取0.25 mL洗脱的phage加入1.75 mL TG1菌液,37 °C水浴45 min。按以下方式稀释涂布计算滴度(空白对照和实验组相同)
|

d) 剩余侵染菌液5000 g离心5 min,去上清,重悬沉淀后铺1块150 mm的TYE(+)平板,37 °C培养过夜。
e) 对滴度检测板计数和计算,对向150 mm板中加入4 mL 2TY刮板,混匀后加入甘油至20%,-80 °C储存或进行下一步制备phage。
f) 接种75 μL刮板液接种至50 mL 2TY/2%G/Amp中,37 °C、220 rpm培养1.5~2 h至OD600为0.4~0.5之间。
g) 取10 mL菌液加入2 μL保存的KM13辅助噬菌体,37 °C水浴45 min。
h) 3500 g离心10 min,弃上清,沉淀重悬至50 mL 2TY/0.1%G/Amp/Kan中,25 °C、220 rpm培养16 h。
i) 3500 g、4 °C离心30 min,取40 mL上清加入10 mL 20% PEG/NaCl,混匀,冰浴1 h。
j) 3500 g、4 °C离心30 min,加入2 mL PBS重悬,混匀,17900 g、4 °C离心15 min,上清转移至新的离心管中,取800 μL加入200 μL甘油,混匀,取5 μL加入15 μL PBS中混匀,测量OD260值,并根据公式(pfu/mL = OD260 ×100 ×22.14×1010)计算滴度。
2) 第2轮筛选
与第1轮筛选相比主要有如下不同:
A.加入的第1轮洗脱扩增的phage量改为1×1012 pfu。
B. Wash的次数改为20次。
C. 侵染及测量洗脱的phage滴度,按如下方式进行,对照从2中取100 μL(相当于0.2 μL phage)和10 μL(相当于0.02 μL phage)涂板:
D.铺2块150 mm TYE(+)平板扩增。
 |
3) 第3轮筛选
与第2轮筛选相比主要有如下不同:
A. 加入的第2轮洗脱扩增的phage量为1×1012 pfu。
B. 测量洗脱的phage滴度按下列方式进行,空白对照从管1中取100 μL(相当于0.2 μL phage)及10 μL(相当于0.02 μL phage)涂板:
C. 第2天取其中2×108 pfu洗脱的phage侵染8 mL TG1,铺2块150 mm TYE(+)平板扩增,并按以下方式测量侵染实际滴度。

D. 刮板后接种75 μL刮板液制备phage,最终用50 mL 2TY/Amp/Kan/0.1%G培养16 h,取40 mL制备phage。
4) 更多轮筛选。
如果第3轮筛选得到的单克隆phage ELISA结果不理想,可进行第4轮及更多轮筛选,方法同第3轮。
4.多克隆phage ELISA检测筛选结果
1) 包被抗原
A.分别取抗原0.2 μg/孔(总体积为100 μL,溶于GFBE),另外设一个空白对照,分别加入96孔免疫板中。
B.放置4 °C过夜(16 h)包被。
2) 封闭
A. GFB洗2次。
B. 每孔加入300 μL 5%牛奶室温放置3 h,完成封闭。
3) 孵育1抗
A. 分别取1×1011 pfu的原始文库、第1、2、3轮筛选扩增的phage至420 μL 5%牛奶中混匀。
B. GFB洗2次。
C. 向每组的孔中分别加入100 μL 上二步(A)中的phage,置于100 rpm的平台上室温孵育1 h。
D. GFBT洗5次。
4) 孵育2抗
A. 取适量HRP-M13抗体,用5% 牛奶1:8000稀释。
B. 加入100 μL到上面的各孔中,置于100 rpm的平台上室温孵育1 h。
C. GFBT洗4次。
5) 显色
A. 在避光条件下,每孔各加入100 μL TMB,室温孵育5 min至阳性孔蓝色较深。
B. 每孔各加入50 μL 1 M H2SO4终止反应,此时溶液呈现黄色。
C. 采用酶标仪读取OD450值。
5. 单克隆phage ELISA筛选阳性克隆
1) 制备单克隆phage
A. 取96孔细胞培养板,每孔加入100 μL 2TY/2%G/Amp。
B. 分别在phage侵染计数的平板上挑单克隆接种至96孔细胞培养板中。37 °C、250 rpm培养6~8 h。
C. 另取一块96孔细胞培养板,分别加入200 μL 2TY/2%G/Amp,分别取5 μL以上菌液接种到对应的孔中。37 °C、250 rpm培养1.5 h。
D. 取8 μL KM13至1 mL 2TY中混匀,分别向以上各孔中加入5 μL KM13稀释液。37 °C静置45 min。
E. 采用排枪混匀并吸走150 μL菌液,剩余菌液3500 g离心15 min,甩掉上清,在纸巾上倒置2 min。
F. 每孔加入200 μL 2TY/0.1%G/Amp/Kan,25 °C、250 rpm培养16 h。
G. 取80 μL 500 mM EDTA到5 mL PBS中混匀,用排枪取50 μL/孔加入一块新的96孔板中。
H. 将上两步(步骤F)的菌液3500 g离心30 min,各取150 μL上清转移至上一步(G步)的96孔板,4 °C储存备用。
2) 包被抗原和封闭
方法同上
3) 孵育1抗
取以上50 μL上清加入200 μL 5%脱脂奶粉混匀后作为1抗孵育,其它同上。
4) 孵育2抗同上
5) 显色同上
6. 测序分析比对阳性单克隆
7. 单克隆可溶性ELISA检测阳性克隆
1) 抽提阳性非重复单克隆质粒被转化HB2151感受态
A. 取50 μL保种的单克隆TG1菌接种5 mL 2TY/Amp/2%G,37 °C培养12 h。
B. 保种后,各取4 mL菌液抽提质粒。
C. 转化HB2151感受态细菌,涂布TYE(+)平板。
2) 制备单克隆可溶性抗体
A. 分别挑取1个单克隆接种至100 μL 2TY/Amp/2%G,于96孔板中,37 °C、250 rpm培养8~10 h。
B. 各取5 μL菌液重新接种200 μL 2TY/Amp/2%G中(剩余菌液各加入100 μL 30%甘油后-80°C冻存),于96孔板中,37° C、250 rpm培养2小时。
C. 3500 g离心10 min,甩板以弃去上清,每孔各加入200 μL 2TY/Amp/IPTG(1 mM),25 °C、250 rpm培养16 h。
D. 取80 μL 500 mM EDTA到5 mL PBS中混匀,用排枪取50 μL/孔加入一块新的96孔板中。
E. 将上2步(C)的菌液3500 g离心30 min,各取150 μL上清转移至上一步(D步)的96孔板, 4 °C储存备用。
3) ELISA检测
A. 包被抗原:同上
B. 封闭:同上
C. 孵育1抗:取以上50 μL上清,加入200 μL 5%脱脂奶粉混匀后作为1抗孵育,其它同上。
D. 孵育2抗:取HPR标记的抗myc抗体用5%牛奶进行1:4000稀释后(终浓度约0.2 μg/mL)孵育,其它同上。
针对性建议(TROUBLESHOOTING)
1. 噬菌体展示筛选的方法有很多种,这里介绍的是其中经典的一种,即在免疫管直接包被靶标蛋白筛选。
2. 筛选过程中可以适当提高第3轮以后的Tween 20的浓度,如0.2~0.4%,从而筛选高亲和力的抗体。
3. 决定噬菌体筛选能否获得搞亲和力抗体的最关键因素是文库质量。
4. 具体需要筛选几轮要根据多克隆ELISA及单克隆ELISA结果而定。
5. 这里采用的是胰蛋白酶消化结合的噬菌体,对于其他文库不一定适合。
3.3.3 配体指数富集的系统进化技术筛选核酸适配体
简介(INTRODUCTION)
SELEX(systematic evolution of ligands by exponential enrichment.A technique is described for the identification of nucleic acid sequences bound with high affinity by proteins or by other molecules suitable for a partitioning assay. Here, a histidine-tagged protein is allowed to interact with a pool of nucleic acids and the protein–nucleic acid complexes formed are retained on a Ni-NTA matrix. Nucleic acids with a low level of recognition by the protein are washed away. The pool of recovered nucleic acids is amplified by the polymerase chain reaction and is submitted to further rounds of selection. Each round of selection increases the proportion of sequences that are avidly bound by the protein of interest. The cloning and sequencing of these sequences finally completes their identification.
材料(MATERIAL)
r 试剂(REAGENTS)
1. Taq polymerase、50 bp marker、Absolute ethanol、95% ethanol、Phenol: Chloroform (1:1)、3 M NaOAc of pH 5
2. Synthetic oligonucleotide library
3. Primers (T7 promoter as forward and t7 terminator as reverse primers)
4. Partitioning matrix (NTA-agarose (Ni2+ beads, Qiagen))
5. pCR-2.1-TOPO vector (Invitrogen) for TA cloning
r 实验前准备(SETUP)
1. Binding buffer: Binding buffer (BB):50 mM Tris–HCl pH 7.5、 150 mM NaCl, 20 mM KCl、1 mM DTT、0.05% NP 40、1 mM MgCl2、2.5% polyvinyl alcohol (PVA)、1 mM EGTA、50 µg/mL poly(A)、2 μL/mL vanadyl ribonucleoside complex (VRC)、0.5 µg/mL tRNA、125 µg/mL BSA.
2. Binding buffer :20 mM HEPES pH 7.5、150 mM NaCl、5 mM MgCl2、and 5% glycerol
3. Washing buffer :A 200 mM KCl、20 mM HEPES pH 7.4、5 mM MgCl2、5% glycerol
4. Elution Buffer: 20 mM Tris HCl pH 8.0、250 mM NaCl、 500 mM imidazole
r 器械(EQUIPMENT)
核酸电泳系统、离心机
实验步骤(PROCEDURE)
1. Synthesize an oligonucleotide
A 60 bp long DNA oligo containing a T7 promoter, a random 20 mer sequence and followed by a T7 terminator was synthesized by IDT (Fig. 1). It was used as a template to amplify a library by polymerase chain reactions (PCR).
 |
2. PCR Optimization
PCR Conditions
Component | Volume per Reaction | I. Conc. | F. Conc. |
10× PCR Buffer | 2.5 μL | 10× | 1× |
MgCl2 | 0.5 μL | 50 mM | 1 mM |
dNTP Mix | 0.5 μL | 10 mM | 0.2 mM |
For Primer | 5 μL | 10 μM | 2 μM |
Rev Primer | 5 μL | 10 μM | 2 μM |
Taq DNA Pol. | 0.25 μL | 5 U/μL | 0.05 U/ μL |
Template/NTC2 | 1 μL | 100 nM | 4 nM |
ddH2O | Up to 25 μL |
|
|
Total Volume (μL) | 25 μL | -- | -- |
PCR program
Temperature | Time | Round | Cycles |
99°C | lid temperature |
|
|
95°C | 5 min |
|
|
95°C | 30 sec |
|
|
5°C | 30 sec |
|
|
72°C | 30 sec | 2 | 10 (8-25) |
72°C | 5 min |
|
|
4°C | Hold |
|
|
Purify the PCR product and Elute PCR product with 50 μL Elution buffer. The quality of the double stranded random DNA should be examined by 4% agarose gel electrophoresis and Nanodrop of OD260.
▲Store the DNA pool at − 20°C.
3. Preparation of the partitioning matrix
1)Take 50 μL of Ni-NTA agarose beads (Qiagen), and wash it twice with 1 mL of sterile water or Ni Binding buffer to remove all storage buffers (by a 30 sec 8000 g centrifugation in a bench top centrifuge).
2)Wash third time and remove 800 μL of buffer.
4.Attachment of purified protein
1)Add about 3 mg of purified histidine-tagged protein into tube. Incubate 1 hr at 4 ˚C with rocking on a roller to allow binding of the protein on the Ni-NTA beads.
2) Centrifuge tube for 30 sec at 8000 g remove supernatant, and then wash the beads twice with 500 mL of Ni Binding buffer to remove all unbound protein.
3)Mix the PCR purified product (50 μL) in 200 μL low salt (Ni Binding buffer) buffer (100 mM KCl, 20 mM HEPES pH 7.4) containing 50 μg/mL primer DNA.
4)Incubate the mixture (protein + oligo) for 1 hr at 4˚C with rocking for stability of the complex
5.Washing with Binding buffer
 |
Wash the beads three times with 500 mL of Ni Binding buffer (20 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM MgCl2, and 5% glycerol) each time. Optional step: Fourth time wash with Ni Binding buffer containing additional concentration KCl i.e. 200 mM (200 mM KCl, 20 mM HEPES pH 7.4).
6.Elution and purification of protein-DNA complex
1)Use 200 μL Elution buffer (20 mM Tris HCl pH 8.0, 500 mM NaCl and 500 mM imidazole) to elute the protein-DNA complex. Mix well by inverting the tube few times followed by centrifugation of 30 sec 8000 g
2)Pool the complex fractions into new 1.5 mL tube.
7.Extraction of DNA from complex
1) Add 100 μL of sterile water and 200 μL of phenol: chloroform (1:1). Vortex 30 sec and spin 5 min at full speed (21,000×g). Recover the upper aqueous phase.
2) Repeat this extraction once more time.
3)Add 2 μL of 1 M MgCl2, 20 μL of 3 M NaOAc of pH 5, and 700 μL of 100% EtOH. Precipitate for at least 30 min at −20°C; Spin at 30 min at 21,000 × g, carefully pour out the supernatant (it’s OK to leave a little supernatant behind).
4)Wash the pellet with 500 μL-1 mL of 70% EtOH, vertex it and spin 30 min at 21,000 × g.
5)Carefully Pour out the supernatant, dry the pellet under vacuum dissector for 5 min and re-suspend it in 13 μL of sterile water.
8.Amplification of eluted DNA
1)Amplify the eluted DNA and run on 4% agarose gel and purify by a PCR purification kit.
2)Repeat the cycles (2. to 8.) and saturate the target oligonucleotide pool. The purified DNA will be further used as a template and amplified with similar protocol as above.
9.Do a second round of selection and amplify the eluted DNA
10.Do a third round, or more rounds of selection if necessary.
11Finally, screen the eluted protein-DNA1 by gel filtration S200-column in high salt buffer (250 mM NaCl, 20 mM Tris-HCl pH 8.0). The protein-DNA complex will migrate into earlier peak as compared to protein alone sample, while the majority unbound DNA will elute in later peaks, which indicates some DNA form a complex with the protein. Collected the fractions should be checked on 6% native polyacrylamide gel and purify it. The purified DNA is called protein-DNA1.
12.Cloning and Ligation
1)Set up the TOPO® cloning reaction (mix together selected DNA and pCR4-topo vector called lmo-DNA2).
2)Incubate for 5 minutes at room temperature • Transform the TOPO® cloning reaction into One Shot® Competent Cells or equivalent.
3)Select and analyze 10 white or light blue colonies for insert.
4)Sequence the clones; or you might want to do a deep sequencing of the selected DNA to find out the sequences.
13.Analyze sequencing data
1)Sequencing of the DNA pool produces both sense and antisense strands. To analyze the data, separate sense strands from antisense strands. To accomplish this, follow Steps (2)–(9), as detailed below.
2)Save the data using two file names, such as ‘sense’ and ‘antisense’.
3)Using the sense file, search all sequences containing the forward primer and color code them.
4)Delete any sequences without the forward primer code.
5)Remove both the forward primer from the 5′ end of the sequences and the reverse primer at the 3′ end, leaving only the initial randomized region.
6)Repeat Steps (3)–(5) for the antisense.
7)Change all antisense sequences (Step (6)) to the complementary strand using the Integrated DNA Technologies website.
8)Combine all these sequences together and determine the sequence alignment using sequence alignment programs such as ClustalX1.83.
9) Group sequences into families.
 |
3.4 检测蛋白与互作分子的互作
3.4.1 酵母双杂交 Yeast two hybrid
简介(INTRODUCTION)
酵母双杂交系统(yeast two-hybrid system)是在酵母体内分析蛋白质-蛋白质相互作用的基因系统,也是一个基于转录因子模块结构的遗传学方法。酵母双杂交由Fields在1989年提出,它的产生是基于对真核细胞转录因子特别是酵母转录因子GAL4性质的研究。GAL4包括两个彼此分离的但功能必需的结构域:位于N端1-147位氨基酸残基区段的DNA结合域(DNA binding domain, DNA-BD)和位于C端768-881位氨基酸残基区段的转录激活域(Activation domain, AD)。DNA-BD能够识别位于GAL4效应基因GAL4-responsive gene的上游激活序列(Upstream activating sequence, UAS),并与之结合。而AD则是通过与转录机构(transcription machinery)中的其他成分之间的结合作用,以启动UAS下游的基因进行转录。DNA-BD和AD单独分别作用并不能激活转录反应,但是当二者在空间上充分接近时,则呈现完整的GAL4转录因子活性并可激活UAS下游启动子,使启动子下游基因得到转录。
为了验证两个蛋白相互作用,我们将蛋白A与Gal4的DNA-BD融合(将蛋白A的DNA克隆至能表达DNA-BD融合蛋白的质粒中,如pGBKT7,该质粒能表达Trp),而蛋白B与AD融合(将蛋白B的DNA克隆至能表达AD融合蛋白的质粒中,如pGADT7,该质粒能表达Leu),质粒共同或依次转化进入酵母细胞AH109后,如果蛋白A能与蛋白B结合,则会启动下游报告基因ADE2、HIS3和MEL1的表达,表达产物分别为Ade、His和α-半乳糖苷酶。AH109本身不能在缺少Leu、Trp、Ade、His的培养基中生长,也不能表达α-半乳糖苷酶而在X-α-Gal平板变蓝。因此当同时转化了蛋白A蛋白B质粒后,如果某些AH109克隆能在SD/-Trp/-Leu/-Ade/-His/X-α-Gal的培养基中生长并变蓝的话,则可推测该克隆中的蛋白B能与蛋白A结合,该克隆视为阳性克隆,经过下游假阳性分析,最终证明两个蛋白相互作用。见下图:

我们实验室用的酵母菌菌株是AH109,它含有4个报告基因,如下图:
HIS3报告基因启动后,酵母可以在组氨酸确实的平板上生长;ADE2报告基因启动后,酵母可以在腺嘌呤(adenine)缺失的平板上生长;lacZ和MEL1报告基因是平行的,两个基因上游都是由MEL1 UAS和MEL1 TATA区域同时控制的,区别在于lacZ的产物是β-半乳糖苷酶,其底物是X-gal;而MEL1的产物是a-半乳糖苷酶,其底物是X-a-gal。但实际实验中如果以X-gal代替X-a-gal是无法得到蓝色阳性克隆的,因此筛选的培养基中需要加X-a-gal作为显色底物。由这三种UAS共同调控的AH109系统可以较好地排除假阳性,并且也能检测结合强弱。以下是不同培养基所能检测出的结合强度:
培养基 | 结合强度 |
-L/-W/-H/-A/X-a-gal | ★★★ |
-L/-W/-H | ★★ |
-L/-W | ★ |
材料(MATERIAL)
r 试剂(REAGENTS)
YPDA液体培养基/YPD液体培养基(不加adenine,其他都一样)、各种类型的选择性培养基、两缺(LW+ adenine)平板、三缺(LWH+adeninel)平板、四缺(LWH-adenine±X-gal)平板、10× TE buffer、10× LiAc buffer、1 M LiAc溶液,高压灭菌。、50% PEG 3350 (w/v)、1.1×TE/LiAc buffer、1× TE/LiAc/PEG3350 buffer、50% 葡萄糖(w/v)、10 mg/mL ssDNA (salmon sperm DNA)
r 实验前准备(SETUP)
高压灭菌50 mL离心管、1.5 mL Ep管、水、以及所有相关试剂。ssDNA要在100°C金属浴灭活10分钟后,放在冰上,待用。无菌96孔板(LWH板上筛选时需要用)
r 器械(EQUIPMENT)
Thermo离心机(不需要预冷,常温即可)、金属煮样器、台式离心机、30°C摇床、30°C培养箱、超净台
实验步骤(PROCEDURE)
1. 活化酵母 ►预计时间消耗:2~3天用接种环将酵母AH109从甘油冻存菌中挑一点,在YPDA平板上划线,并在30 °C培养箱中培养2~3天进行活化。
2. 摇过夜菌 ►预计时间消耗:一晚将活化的酵母中挑一个较大的单克隆至10 mL YPDA培养基中,在30 °C摇床中摇过夜。
▲ 关键步骤:50 mL离心管的管盖不要拧得太紧。挑一个较大的克隆之后,在涡旋仪上进行充分地涡旋,有助于酵母的生长。
3. 制备感受态►预计时间消耗:30分钟
1) 每100 mL酵母可以做12管感受态。这里我们以100 mL为例进行介绍。
2)将0.3 mL过夜酵母菌加入至100 mL YPDA中(保证此时的OD600在0.1左右),30 °C摇床中培养。
3)待OD值达到0.4 ~ 0.6之间时,将100 mL菌液均分到两个50 mL离心管,1500 g离心5分钟。
4)弃上清,用30 mL灭菌水重悬,并混到一管中。离心1500 g离心5分钟。
5)弃上清,用1 mL 1×TE/LiAc buffer重悬,转移至无菌1.5 mL Ep管, 12000 g离心15 s。
6)弃上清,用600 μL 1×TE/LiAc buffer重悬,放在冰上,待用。
▲关键步骤:离心机不需要预冷,常温即可。制备好的感受态放在冰上,在1小时内使用。
4.转化►预计时间消耗:1.5小时
1)在1.5 mL Ep管中,标记好BD和AD名称,向每管加入AD和BD各100 ng,再加入5 μL灭活的ssDNA,随后加入50 μL的酵母感受态,在涡旋仪上涡旋混匀。
2)每管加入500 μL 1×TE/LiAc/PEG 3350 buffer,在涡旋仪上混匀后,在30 °C摇床孵育30分钟。每隔10分钟轻轻弹动Ep管使菌液混匀。
3)孵育完,每管加20 μL DMSO,在42 °C水浴锅中孵育15分钟,每隔5分钟轻轻弹动Ep管使菌液混匀。
4)在台式离心机12000 g离心15 s收集酵母,弃上清,加入1 mL YPD培养基重悬,在30 °C 摇床孵育1小时(这段时间可以随便调整,不需要严格1小时)。
5)在台式离心机12000 g离心15 s收集酵母,弃上清,用500 μL 0.9% NaCl溶液重悬,再次离心12000 g离心15 s,收集酵母,弃上清,用100 μL 0.9% NaCl溶液重悬,全部加到LW平板上涂板,并在30 °C培养箱培养3天。
▲关键步骤:
♫ LW平板要提前放在30 °C,使表面的水分尽量蒸发掉,不然涂板时琼脂无法很快地吸收菌液。
♫ AD和BD的量在100-400 ng都可以,但要记住:质粒含量越多对酵母的毒性也越大,这会影响转化效率。转化后加YPD培养基孵育过程很重要,这一步可以大大提高转化效率。
♫ 转化过程中的步骤(5),必须用NaCl清洗一遍,防止有少量YPD残留导致LW平板上长出非目的克隆。
5. 筛选相互作用►预计时间消耗:3天
1) 在无菌的96孔板上,用排枪加无菌水,每孔100 μL。
2) 将LW平板上的克隆挑单克隆,在对应的孔中吹打数次,将酵母菌溶解在水中。
3) 每个LW平板上至少挑3个单克隆,做平行对照。
4) 将repeater(所谓盖章用的章)用酒精棉球擦拭,待酒精挥发后,在酒精灯上烧,放在边上晾凉。
5) 在LWH板和LW板上标记好,把repeater放在96孔板上,沾酵母,平行移到对应的平板上“盖章”。
6) 待菌液完全被平板吸收后,倒放置在30 °C 培养箱,培养3天。
针对性建议(TROUBLESHOOTING)
1. 实验前先明确实验目的,是要验证相互作用,还是要验证不相互作用。如果是验证相互作用,那么在选择筛选平板的时候要选择结合强度最高的,如LWH+X-a-gal的平板,这个平板是AH109的3个reporter基因全部表达之后酵母才能生长,否则少一个都长不了,或者变不了蓝色。如果是验证不相互作用,那么就要选择弱筛选培养基,如-L/-W/-H/+A培养基,在这么弱选择性的培养基上都无法生长,那绝对是没有相互作用的。
2.做实验的过程尽量不要混杂细菌实验,这样很容易被细菌污染,长出来的盘子上,淡黄色的是细菌,纯白色的是酵母。
3.4.2 双分子荧光互补Bimolecular fluorescence complementation
简介(INTRODUCTION)
GFP的发光机制:
|

GFP由238个氨基酸分子组成,分子量为26.9 kDa。来源于水母的野生型GFP在395 nm和475 nm分别有主要和次要的激发峰,它的发射峰在509 nm,处于可见光谱的绿色区域;来源于海肾的GFP只在498 nm有单个激发峰。
GFP是典型的β桶形结构,包含β折叠和α螺旋,将荧光基团包含在其中。严密的桶形结构保护着荧光基团,防止它被周围环境猝灭,内部面向桶形的侧链诱导Ser65-Tyr66-Gly67三肽环化,导致荧光基团形成。
|

双分子荧光互补机制:Hu等在2002年最先报道了一种直观、快速地判断目标蛋白在活细胞中的定位和相互作用的新技术。在GFP的两个β片层之间的loop环结构上有许多特异位点可以插入外源蛋白而不影响GFP的荧光活性,该技术巧妙地利用该荧光蛋白分割成两个不具有荧光活性的分子片段(N-fragment,C-fragment)。这些片段在体内体外混合时,不能自发地组装成完整的荧光蛋白,也不能激发出荧光。这2个荧光蛋白的片段分别连接融合两个目标蛋白,如果荧光蛋白活性恢复则表明两目的蛋白发生了相互作用,在空间上互相靠近互补,重新构建成完整的具有活性的荧光蛋白分子,在该荧光蛋白的激发光激发下发射荧光。反之,若蛋白之间没有相互作用,则不能被激发出荧光。
BiFC技术研究进展
近几年,BiFC技术迅速发展,Hu和Kerppola系统地研究了GFP及其3个不同颜色的突变体蓝色荧光蛋白BFP,青色荧光蛋白CFP和黄色荧光蛋白YFP的BiFC现象。随后发展出的多色荧光互补技术(muticolor BiFC)不仅能同时检测到多种蛋白复合物的形成,还能够对不同蛋白间产生相互作用的强弱进行比较。如多色双分子荧光互补,三分子荧光互补,BiFC-FRET等。该技术已用于转录因子,G蛋白βγ亚基的二聚体形成,不同蛋白间产生相互作用强弱的比较以及蛋白泛素化等方面的研究工作上。
BiFC技术的优缺点
优点:
适用于体内和体外蛋白相互作用的研究。
在细菌,真菌以及真核细胞中检测时,所研究的蛋白处于天然环境中,并且能直观报道蛋白相互作用在细胞中的定位研究。
BiFC技术耗时较短。
相对于FRET和BRET技术对仪器的要求高,数据处理复杂,而BiFC只需要荧光倒置显微镜,数据处理简单,只是检测荧光的有无,背景干净,检测更加灵敏,不依赖于其他次级效应。
BiFC技术还可以用于研究蛋白之间的弱相互作用或瞬间相互作用。
不需要蛋白有特别的理论配比,能检测到不同亚群蛋白间的相互作用。
缺点:
对系统温度敏感,温度太高,片段不易互补形成完整的荧光蛋白。一般在30 °C以下形成互补效应好,温度越低,越有利于片段的互补,这对细胞在生理条件下的蛋白相互作用带来不利因素。
观察到的双分子荧光信号滞后于蛋白的互作过程,不能实时观察蛋白相互作用或蛋白复合物的形成过程。
材料(MATERIALS)
r 试剂(REAGENTS)
转染试剂(Lipo2000(Invitrogen),Lipo6000(全式金))、PBS、3.7%甲醛(避光)、DAPI(+抗淬灭剂), UltraCruz,mounting medium,sc-24941、指甲油、载玻片,用酒精或酸洗干净,烘箱烘干
r 实验前准备(SETUP)
以细胞为例,培养生长状态较好的细胞
r 器械(EQUIPMENT)
荧光正置显微镜、镊子、自制弯曲的1 mL注射器针头、Microscope cover glass 18 mm×18 mm (NEST)、24孔培养板
实验步骤(PROCEDURE)
1. 将目的基因插入到含有N片段或C片段的载体中,构建成融合蛋白表达载体。两种蛋白需要互换载体。N-fragment将目的蛋白插入C端,而C-fragment将目的蛋白插入到N端。
2.前日传代的HeLa cell/293T,吸取培养液,PBS洗两次,轻柔摇小皿,加入200 μL胰酶37 °C消化2 min,之后有大片细胞团飘起时,加入1 mL DMEM终止消化反应,快速但轻柔吹吸细胞,使其悬浮。取100 μL用于计数。
3. 细胞铺片:
1) 用镊子夹起cover glass边缘,在酒精灯上过一下,竖立放入24孔板中。
2) 每孔加入450 μL DMEM,用枪头轻轻抵一下玻片,使其浸润,确保底部没有气泡。
3) 向每孔加入约20 μL消化下来的细胞(这个依据细胞量,一般爬片细胞尽量少分细胞,呈现单个细胞有利于成像观察,一开始可以更少分,显微镜下观察细胞的量,再逐渐增多。),37 °C培养。
▲关键步骤:用于荧光检测的细胞,要求底部呈单细胞铺片,因此少分一些细胞,大约0.2~0.5×105/ mL。
4. 次日,细胞约爬满70~80%时进行转染,转染量为0.8~1.0 μg plasmids/孔;两种质粒以1:1加,即各加入0.4~0.5ug/孔;6小时后换液。
5. 转染24~48 h后,吸去DMEM培养液,加入3.7%甲醛(新鲜的配置,避光,放置不可多于一个月),固定8~10 min,37 °C。
6. PBS洗,5 min X 2。
7. 扣片:用1 mL注射器针头(尖端弄弯),挑出铺片的cover glass,放置在铺有封口膜的24孔盖子上的PBS里,铺有细胞的一面朝上。
8. 镊子夹起玻片边缘,滤纸轻轻擦拭吸去PBS,不可碰到细胞。
9. 载玻片上,远离磨砂边缘处,滴1~2 μL DAPI(含有抗猝灭剂),将玻片倒扣在载玻片上。
10.指甲油封片,室温晾干,一般需要涂抹2~3次,才能完全封住。约30 min,之后避光保存在-20 °C冰箱,待观察。
11.荧光倒置显微镜下观察蛋白的相互作用。488 nm激发的绿色荧光团为检测目标,同时紫外线激发的蓝色荧光标记细胞核,适合的物镜倍数是20~40X。
附:应用实例

针对性建议(TROUBLESHOOTING)
1. 荧光片段和目标蛋白之间最好加一个连接肽,以避免蛋白空间位阻所导致的片段间不能相互靠近。常用的连接肽氨基酸序列有RSIAT,RPACKIPNDLKQKVMNH和GGGGS等。
2. 温度对片段间互补影响很大,可以有两种解决方案。一是在室温或低于室温(≤30 °C)下培养细菌或细胞,二是在生理条件下培养细菌或细胞,使融合蛋白正常表达,然后将培养物低温处理1~2 h或接着室温培养1 d;本实验室所构建的V124和V125BiFC载体,温度对其没有影响,大家可以放心正常使用。
3. 建立阴性对照。以便更加确信BiFC信号反映的是蛋白质之间的相互作用。阴性对照通常是将相互作用的蛋白进行突变,降低或缺失其相互作用能力,再采用相同策略的BiFC系统检测。
3.4.3 Western Blotting
简介(INTRODUCTION)
这种技术是把电泳分离的组分从凝胶转移到一种固相支持体(NC、PVDF膜),并以特异性抗体作为探针检测。抗体与附着于固相支持体的靶蛋白所呈现的抗原表位发生特异性反应。这种技术的作用是对非放射性标记蛋白组成的复杂混合物中的某些特异性蛋白进行定性、分析。
材料(MATERIALS)
r 试剂(REAGENTS)
20% Tween20、10 mM PMSF、0.2 M NaH2 PO4、0.2 M Na2HPO4、TBST、转移膜、化学发光试剂、转移缓冲液(TB buffer)、电泳缓冲液(EB buffer)、PBS 缓冲液、10×丽春红染液、封闭液(5% 脱脂牛奶)、滤纸、SDS-PAGE及其上样缓冲液
r 器械(EQUIPMENT)
垂直板电泳转移装置、高速离心机、分光光度计、脱色摇床、搪瓷盘、蛋白电泳装置、匀浆器
实验步骤(PROCEDURE)
1. 蛋白样品制备:预计时长:1 h
1) 贴壁细胞总蛋白的提取:
A.按1 mL裂解液加10 μL PMSF(100 mM)混匀后置于冰上。
B. 吸去细胞培养基,用4 °C预冷的PBS洗两次后,根据细胞数目加入相应体积的裂解液摇匀于冰上裂解30 min(60 mm 皿加 1 mL)期间可摇动混匀。
C. 裂解完成后,用枪头将细胞刮掉然后转移到1.5 mL EP管中。
D. 2~3步也可为:加入裂解液静置两分钟后刮下细胞,转移到1.5 mL EP管中,用超声破碎细胞。
E. 12000 g×5 min 4 °C离心后,转移至新的EP管中。
F. -20 °C保存或进行下一步。
2) 悬浮细胞总蛋白的提取
A. 将培养基倒至15 mL离心管中,800 g×5 min 4 °C离心,吸出培养基,加入2 mL 4 °C预冷PBS重悬细胞后再次800 g×10 min 4 °C离心,尽量吸干PBS,加入裂解液冰上裂解30 min,裂解过程中要经常弹一弹以使细胞充分裂解。
B. 将裂解液与培养瓶中裂解液混在一起12000 g×5 min 4 °C离心,取上清于1.5 mL 离心管中并置于-20 °C保存或进行下一步。
3) 组织中总蛋白的提取
A.将组织块尽量剪碎,加入含PMSF的裂解液于匀浆器中,进行匀浆然后置于冰上。
B. 几分钟后,再次匀浆,置于冰上。如此反复几次使组织尽量碾碎。
C. 裂解30 min 后,将裂解液移至1.5 mL EP管中,12000 g×5 min 4 °C离心,取上清于1.5 mL离心管中并置于-20 °C保存或进行下一步。
2. 蛋白质定量:预测时长:1.5 h
一般选择BCA或者Bradford方法,具体方法见各试剂盒说明书。BCA要求检测波长为 562 nm,Bradford为595 nm。为避免假阳性结果,建议先把lysis buffer加入BCA工作液或考马斯G250混合看是否产生颜色。具体测完蛋白含量后,计算含50~100 ug蛋白的溶液体积即为上样量(一般8 cm宽的迷你胶每个泳道最大能承载的蛋白质量为150 ug)。
1)制作标准曲线
A. 从-20 °C取出1 mg/mL BSA,室温融化后,备用。
B. 取18个1.5 mL离心管,3个一组,分别标记为 0 μg,2.5 μg,5 μg,10 μg,20 μg,40 μg。
C. 按下表在各管中加入各种试剂。
| 0 μg | 2.5 μg | 5 μg | 10 μg | 20 μg | 40 μg |
1 mg/mL BSA |
| 2.5 μL | 5 μL | 10 μL | 20 μL | 40 μL |
0.15 M NaCl | 100 μL | 97.5 μL | 95 μL | 90 μL | 80 μL | 60 μL |
G250 溶液 | 1 mL | 1 mL | 1 mL | 1 mL | 1 mL | 1 mL |
混匀后,室温放置2 min。在生物分光光度计上比色分析。
2) 检测样品蛋白含量
A. 取足量的1.5 mL离心管,每管加入4 °C储存的考马斯亮蓝溶液1 mL。室温放置 30 min后即可用于测蛋白。
B. 取一管考马斯亮蓝加0.15 M NaCl 100 μL,混匀放置2 min可作为空白样品,将空白倒入比色皿中做好的标准曲线程序下按Blank测空白样品。
C. 弃空白样品,用无水乙醇清洗比色皿2次,用无菌水洗一次。
D. 取一管考马斯亮蓝加95 μL 0.15 M NaCl溶液和5 μL待测蛋白样品,混匀后静置2 min后测量。
3. SDS-PAGE电泳预计时长:1 h
取50 μg/孔蛋白上样电泳(上样总体积一般不超过15 μL)
4. 转膜预计时长:3 h
1) 在电泳结束前20 min开始准备转膜所需的东西。转一张膜需准备6张的滤纸(长一般为8.1~8.3 cm×宽8 cm)和1张NC或PVDF膜。切滤纸和膜时一定要戴干净手套,因为手上的蛋白会污染膜。PVDF膜使用前用无水甲醇中浸泡1 min~2 min,NC膜不需要。目的是为了活化PVDF膜上面的正电基团,使它更容易跟带负电的蛋白质结合,做小分子的蛋白转移时多加甲醇也是这个目的。
2) 将玻璃板撬开后,轻轻刮去浓缩胶,用蒸馏水冲洗干净。取出凝胶后可以在右上角裁一个小角以区分上下左右。之后有两种方法供选择,一是按照marker指示,把含有自己感兴趣的蛋白的胶裁下来,二是把整张胶转膜。
3) 在陶瓷盆中倒入TB buffer,放入浸过甲醇的PVDF膜平衡10 min。带上手套,将夹子打开使黑的一面平放转移槽的底座,依次在石墨电极上垫一张海绵垫、叠放3张浸泡过缓冲液的滤纸、刚刚完成电泳的凝胶、PVDF膜(此时可在膜右上角减去一个角,以辨明转膜后的蛋白面与非蛋白面)、和另外3张浸泡过缓冲液的滤纸、另一个海绵垫,合起夹子。注意每叠一层就要用玻璃棒或圆筒试管赶去气泡,膜两边的滤纸不能相互接触,接触后会发生短路。
4) 将夹子放入转移槽槽中,要使夹的黑面对槽的黑面,夹的白面对槽的红面。电转移时会产热,所以TB Buffer要在4 °C预冷,转移槽要放在冰水混合泡沫箱中。的一边放一块冰来降温。一般用60 V 转移2 h或200 mA转1.5 h。
5) 转完后将膜用1×丽春红染液染5 min(于脱色摇床上摇,可回收)。然后用水冲洗掉没染上的染液就可看到膜上的蛋白。使用预染marker话可以看见marker的颜色转印到了膜上,但是凭借这个颜色来判断是否转印完全或转印过头是不可靠的。
5.免疫杂交反应预计时长:1 Day
1) 封闭:将膜用TBST浸湿,移至含有封闭液的平皿中,室温下脱色摇床上摇动封闭1 h。
▲ 关键步骤:注意封闭后不要洗脱,(如果是自己配置的封闭液,最好过滤一下以消除固体杂质)。
2) 一抗孵育:将一抗用封闭液稀释(一般用封闭液稀释即可,如果背景不高用TBST稀释也可以)至适当浓度(可以在5 mL EP管中配置工作液,常用稀释倍数为1:1000,具体按抗体说明来,用后可回收重复用,-20 °C保存,避免反复冻融)。
3) 脱色摇床上洗5 min×5次。
4) 二抗孵育:方法同一抗孵育,但一般二抗稀释比较大1:10000 足够。用TBST在室温下脱色摇床上洗4次,每次5 min。
二抗孵育有两种方法:
A.膜孵:撕下适当大小的一块儿封口膜(也可使用保鲜膜)铺于培养皿盖上,使封口膜保持平整,将抗体溶液加到封口膜上,从封闭液中取出膜,用滤纸吸去残留液后,将膜蛋白面朝下放于抗体液面上,掀动膜四角以赶出残留气泡,放于湿盒中,室温孵育1 h或4 °C过夜孵育。用TBST在室温下脱色摇床上洗6×5 min。
B.配制5 mL抗体稀释液于5 mL EP管中,直接将膜放入,4 °C过夜孵育,用TBST在室温下脱色摇床上洗4×5 min。
6.化学发光预计时长:10 min
化学发光反应:在实验台上铺一张一次性手套,将转移膜放在手套上,蛋白面向上。将化学发光的A液和B液两种试剂在EP管内等体积混合(总体积能够覆盖住膜就行,注意吸完A液后吸B液要换枪头,否则整瓶试剂都将报废),然后均匀滴在PVDF膜的蛋白面,反应1~2 min后(无需避光),将PVDF膜上多余的ECL工作液吸干,凝胶图象分析或X-光片曝光。
7.X-光片曝光预计时长:0.5 h
1) 将转移膜放在保鲜膜上,把另一侧翻过来盖在其上,把保鲜膜固定在片夹上。
2) 在暗室中,将配好的显影液和定影液倒入塑料盆中,打开红色照明灯,取出X光片,剪成比膜稍大,叠放3~4层后放在膜上,不要再移动X光片,关上X片夹,计时2~5 min。(多层胶片的目的是找到最合适的曝光度)
3) 时间完成后,打开片夹,X光片放入显影液,出现明显条带后停止显影,放入定影液,
4) 定影5~10 min至胶片透明。
5) 用水将胶片冲干净,晾干,观察结果。定影液和显影液可回收,避光室温放存,但不宜放太久
▲关键步骤:
♫ 发光液的A、B液一定不能相互污染,吸取时一定要注意换枪头。配制好后应立即使用
♫ 暗室中光线很弱,可暗适应一段时间在开始实验,拿胶片的时候避免用指甲滑到,会有划痕。
针对性建议(TROUBLESHOOTING)
1. 一抗、二抗的稀释度、作用时间和温度对不同的蛋白要经过预实验确定最佳条件。
2. 转膜时要把气泡赶走,PVDF膜不能剪的过小,否则空气容易从膜边缘进去,使得蛋白条带晕开。
3.如果封闭液和抗体稀释液都含5%的脱脂奶粉,封闭之后不用TBST洗,如果封闭液含有而抗体稀释液不用,封闭后可用TBST清洗。
附录: 相关试剂的配制
1.丽春红染液
10×丽春红染液 |
丽春红 S 2 g |
三氯乙酸 30 g |
磺基水杨酸 30 g |
蒸馏水至 100 mL,使用时将其稀释 10 倍。 |
2.转移缓冲液
转移缓冲液(48 mM Tris,39 mM甘氨酸,0.037%SDS,20%甲醇) |
甘氨酸(MW75.07) 2.9 g |
Tris(MW121.14) 5.8 g |
SDS 0.37 g |
甲醇 200 mL,蒸馏水至 1000 mL,溶解后室温保存,次溶液可重复使用 3~5 次 |
3.PBS
0.01 M PBS pH 7.3 |
0.2 M NaH2PO4 19 mL |
0.2 M Na2HPO4 81 mL |
NaCl 17 g |
蒸馏水至 2000 mL |
4.BSA
100 mg/mL 牛血清白蛋白(BSA) |
BSA 0.1 g |
0.15 M NaCl 1 mL |
溶解后,-20 °C保存。制作蛋白标准曲线时,0.15 M NaCl进行100倍稀释成 1 mg/mL,-20 °C保存。 |
5. 封闭液(5% 脱脂牛奶的TBST缓冲液),溶解后 4 °C保存。使用时,恢复室温,用量以盖过膜面即可,一次性使用。如不经常用可每次现用现配所需体积。
6. TBST缓冲液(含0.05%Tween20的TBS缓冲液),混匀后即可使用,最好现用现配。
7. TBS 缓冲液(150 mM NaCl,100 mM Tris-HCl pH 7.5)
3.4.4 免疫共沉淀Co-Immunoprecipitation
简介(INTRODUCTION)
免疫共沉淀(Co-Immunoprecipitation)是以抗体和抗原之间的专一性作用为基础的用于研究蛋白质相互作用的经典方法。是确定两种蛋白质在完整细胞内生理性相互作用的有效方法。
当细胞在非变性条件下被裂解时,完整细胞内存在的许多蛋白质-蛋白质间的相互作用被保留了下来。当用预先固化在 argarose beads上的蛋白质A的抗体免疫沉淀A蛋白,那么与A蛋白在体内结合的蛋白质B也能一起沉淀下来。再通过蛋白变性分离,对B蛋白进行检测,进而证明两者间的相互作用。
这种方法得到的目的蛋白是在细胞内与兴趣蛋白天然结合的,符合体内实际情况,得到的结果可信度高。这种方法常用于测定两种目标蛋白质是否在体内结合;也可用于确定一种特定蛋白质的新的作用搭档。
优点:
相互作用的蛋白质都是经翻译后修饰的,处于天然状态;
蛋白的相互作用是在自然状态下进行的,可以避免人为的影响;
可以分离得到天然状态的相互作用蛋白复合物。
缺点:
检测不到低亲和力和瞬间的蛋白-蛋白相互作用;
两种蛋白的结合可能不是直接结合,而可能有第三者在中间起桥梁作用;
必须在实验前预测目的蛋白,以选择最后检测的抗体,所以若预测不正确,得不到理想的结果,因此方法本身具有冒险性。
|
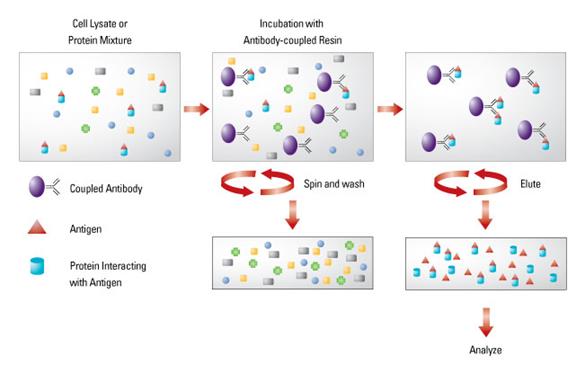
示意图如下:
材料(MATERIALS)
r 试剂(REAGENTS)
IP lysis buffer(100 mM KCl、5 mM MgCl2、0.3% NP-40、20 mM Tris pH 7.5 and Protease inhibitor)
PMSF、PBS、胰酶、Western Blot 所需全部试剂
Beads:strep beads、flag beads、proteinA/G
r 器械(EQUIPMENT)
Thermo 离心机(使用前预冷至4°C)、金属煮样器、旋转台(放于4 °C冰箱中)
实验步骤(PROCEDURE)
使用Beads为SIGMA Anti-Strep ® M2 Affinity Gel为例
1. 从5% CO2,37 °C培养箱中取出转染后24 h-48 h的6孔细胞培养板,除去培养基;用PBS洗两次后,加入约300 μL胰酶,消化1 min后,全部吸入到1.5 mL EP tube中,800 g,离心3 min;再用PBS洗一次,1000 g,离心3 min。
2. 每孔加入300 μL IP lysis buffer(加入PMSF至终浓度为1 mM,注意现配现用。如1 mL IP lysis buffer +10 μL 100 mM PMSF),冰上裂解30 min,之后超声破碎,功率8%,约16 s。
3.12000 g,4 °C、离心10 min。
4.取40 μL上清+10 μL 5×Sample Buffer(20% DTT plus),101°C金属浴10 min,for Input(Western Blot)。
5. 剩余上清全部用来孵育 Beads,4 °C旋转盘4 h或过夜;切记EP tube 要用封口膜封紧,防止盖子开了后,样品就全部毁了。
▲注意:beads使用之前需要用PBS洗3-4次,1000 g,1 min。理论上每孔需要10 μL beads沉淀,例如本实验有三个样,故最后一次加入约600 μL PBS,减掉黄色枪头尖端,平均分为三管(如果全部沉淀的话,很难吸出beads),1000 g离心2 min后,可以看到每管约10 μL的beads白色沉淀,去上清PBS后,分别加入sample lysis。
6.孵育好后,4 °C,8000 RPM 离心1 min,弃上清,加入500 μL IP lysis buffer 来回混匀,切记轻柔。
7.重复步骤(6)5次。
8. 4 °C,8000 RPM 离心1 min,弃上清,加入50 μL IP lysis重悬(为了防止枪头损失beads和样品,不需要移液枪重悬,手指弹即可),加入12.5 μL 5×SDS loading buffer (20% DTT plus),101 °C金属浴10 min,for IP(Western Blot)。
9.制好的Input和IP蛋白样,记录好上样顺序,通过WB进行检测,实验结果如下图所示。

针对性建议(TROUBLESHOOTING)
1. 细胞裂解采用温和的裂解条件,不能破坏细胞内存在的所有蛋白-蛋白相互作用,多采用非离子变性剂(NP40或TritonX-100,本实验购买碧云天的专门用于WB及IP的lysis)。每种细胞的裂解条件是不一样的,通过经验确定。另外细胞裂解液中要加各种酶抑制剂,如商品化的cocktailer,本实验使用的IP lysis buffer需要加入PMSF,现配现用。
2. 本实验步骤适合于已经包被好的strep 或flag beads,很多时候需要用protein A/G预先结合在argarose beads上,使之与含有抗原的溶液及抗体反应后,beads上的protein A/G就能达到吸附抗原的目的。通过低速离心,可以从含有目的抗原的溶液中将目的抗原与其他抗原分离。即清洗完beads后,提前需要将适量抗体(约2 μg,对照组用IgG)加入EP 管,4 °C旋转混匀孵育2 h,瞬时离心弃上清,再清洗beads 3~4次,放4 °C备用。其他操作如上。
3. 在免疫共沉淀实验中要保证实验结果的真实性,各种阳性阴性对照要设置好,应注意:
1) 确保共沉淀的蛋白是由所加入的抗体沉淀得到的,而非外源特异蛋白,单克隆抗体的使用有助于避免污染的发生。
2) 确保抗体的特异性,即在不表达抗原的细胞溶解物中添加抗体后不会引起共沉淀。
3) 蛋白间的相互作用是发生在细胞中,而不是由于细胞的溶解才发生,这需要进行蛋白的定位来确定。
4. 由于蛋白形成复合物以后,某些表位就会被掩盖,因此可能导致使用某一种抗体,无论怎么增加抗体浓度,也极少能将目标蛋白复合物沉淀出来,如有必要最好使用不同抗体分别进行Co-IP。
5. 由于检测的是天然状态,因此在不同的时间和不同的处理下,Co-IP拉下来的蛋白复合物都可能是不同的,增加实验次数,得到的蛋白成员也会更大。
6. Co-IP鉴定得到的蛋白间相互作用可能是直接作用也可能是间接作用,进一步区分还需要进行GST-Pull down等实验检测。
3.4.5 差示扫描荧光技术Differential Scanning Fluorimetry
简介(INTRODUCTION)
原理:差示扫描荧光技术(Differential Scanning Fluorimetry,DSF)是一种快速、经济、高通量的去测量不同条件下配基分子与蛋白结合、稳定蛋白能力的一种方法,当然也可测定突变后蛋白相应能力的改变。原理如下:除膜蛋白外,一般蛋白外表面为亲水氨基酸侧链,内部含有疏水核心,蛋白在活性状态时疏水核心不暴露出来但在变性条件下蛋白失去三级结构导致疏水核心的暴露。而DSF的原理就是荧光染料与蛋白疏水残基非特异的结合程度影响其荧光强度,在荧光染料不与疏水氨基酸侧链结合时其荧光强度弱(极性环境如水导致其荧光的淬灭),与疏水残基结合之后荧光强度增加。当在加热的过程中,蛋白疏水基团暴露导致染料结合逐渐增多从而用来测定蛋白的Tm值、与配基结合的Kd值等。但该方法并不适合用于测定蛋白-蛋白相互作用。该方法具有操作简便、节省样品、高通量等优点,适用于蛋白与配体初步亲和力验证。
材料(MATERIALS)
r 试剂(REAGENTS)
SYPRO orange:200×
r 缓冲液(BUFFER)
150 mM NaCl2、10 mM HEPES pH 7.0
r 器械(EQUIPMENT)
荧光定量PCR仪、RT-PCR板、PCR板掌上离心机
实验步骤(PROCEDURE)
1.将蛋白稀释为不同的浓度并与SYPRO orange染料混合后(注意避光),在荧光定量PCR仪加热(0.1~0.2 °C/s,25 °C~95 °C)并测定荧光读数,用于预测定合适的蛋白浓度(荧光值不能太弱)以进行后继步骤。(具体的参数设置依据仪器说明)
2.使用预测定的浓度的蛋白与不同浓度(对半稀释,8个梯度)的配体混合(三组重复)后加入SYBR orange染料,在荧光定量PCR仪加热(0.1~0.2 °C/s,25 °C~95 °C)并测定荧光度数。以荧光强度做Y轴,温度做X轴作图。可以产生一个“S”形的曲线(图左)用于描述蛋白折叠和去折叠的两种状态的转变,将图1曲线进行求导得到d(flu)/dt与温度的图(图右),由左图可知当“S”曲线斜率既d(flu)/dt最大时对应的温度值为Tm。但不要将此Tm值解读为达到配基结合能力一半时的配基浓度,实际上这时的Tm值可看成蛋白开始发生去折叠时的温度。
|

3.实验结束后将数据导出,可以用Prism作图,拟合得到Tm值。当蛋白结合某些配体或者小分子后,Tm会升高。因此可以用来定性,验证蛋白与小分子的相互作用。
附:数据处理与结果
验证MBP与糖(sucrose、maltose)的结合。
1. 经如上实验步骤获得实验数据,导入Prism中做曲线图,得如下结果:
|

2. 对数据进行拟合求Tm值之前,需要移除一部分原始数据(热力曲线的最低点的前部分和最高点的后部分),之后得如下图:

3. 利用Prism拟合求Tm值(举例MBP +lactose):

4. 最终结果:

针对性建议(TROUBLESHOOTING)
1. 步骤一摸索蛋白最优浓度很重要,不能省略;
2.PCR板上机前需要用掌上离心机瞬离,去除溶液中的气泡。
3.4.6 荧光偏振Fluorescence polarization
简介(INTRODUCTION)
荧光偏振(FP)是一种用于定量分析分子间相互作用的方法。
其基本原理是:标有荧光的分子经单一平面的偏振光(激发光)照射后,吸收光能跃入激发态,随后回复至基态,并发出单一平面的偏振荧光(发射光)。而激发光与发射光的偏振平面偏差的程度与荧光分子的旋转速度呈正相关,与其分子大小呈反相关(既大分子旋转速度会变慢)。
FP反应系统中两种分子无相互作用时,荧光素标记的分子呈游离的小分子状态。由于其分子小,在液相中转动速度较快,测量到的激发光与发射光的偏振平面偏差大,读出的FP值小,反之,如果两种物质有相互作用,形成大分子的复合物,此时检测到的激发光与发射光的偏振平面偏差小(假设分子静止则激发光与发射光的偏振平面重合)读出的FP值大。通过检测反应体系中发射光偏振平面的偏转程度就可以精确地得知两种物质间的结合系数。最适宜检测小至中等分子物质。
|

材料(MATERIALS)
r 试剂
FP buffer(一般是低盐缓冲液,根据自己实验需要配制,此例为5% glycerol, 80 mM NaCl,2 mM MgCl2,20 mM Tris-HCl pH 8.0)、锡箔纸
r 器材
带偏振光模块的酶标仪、黑色96孔板、EP管、PCR管、PCR仪
实验步骤(PROCEDURE)
这里只介绍蛋白-短双链DNA相互作用的实验方法.
1. 订购一端带有荧光分子标记的ssDNA分子(如此例为5’端6-羧基荧光修饰)及其互补链(无修饰),注意只需单链标记即可,不必两条核酸链都标记。
2. 用合适的缓冲液或去离子水溶解单链DNA探针及其互补链至200 μM(为避免荧光分子长时间暴露在光线下发生淬灭,在此步骤及之后的操作中应注意避光)。
3.退火反应获取双链DNA。在PCR管中对照下面表格建立退火体系(避光)。
Samples | Final concentration | Stock concentration | Volume (20 μL) |
5´-6-FAM probe ssDNA | 50 μM | 200 μM | 5 |
complementary strand ssDNA | 50 μM | 200 μM | 5 |
annealing buffer (10×) | 1× | 10× | 2 |
deionised water |
|
| 8 |
Annealing buffer (10×):100 mM Tris pH 8.0,500 mM NaCl,10 mM EDTA
反应混合物在PCR仪器运行退火反应程序:95 °C,2分钟,45-60分钟内缓慢降温至25 °C,样品瞬时离心后置于4 °C储存。
4. 可选。退火反应产物检测依据目的产物的分子量大小选择合适浓度的native PAGE进行电泳,检测反应产率及产物纯度。
5. 探针浓度的确定将欲探究的核酸(双链或和单链DNA探针)用1×FP buffer进行稀释,首先获得60 μL终浓度500 nM的探针,再取出30 μL按照体积比1:1减半稀释7次,得10个终浓度分别为500、250、125、62.5、31.25、15.625、7.8125、3.90625、1.953125和0 nM的探针各30 μL。每个浓度的探针各取20 μL于洁净的黑色96孔板中(注意不要加入气泡),用酶标仪在荧光偏振模式下测量荧光探针的偏振情况(此处用绿色偏振滤片,标准检测速度,增益50)。取平行偏振光强度对探针浓度作图,观察探针浓度与平行偏振光强度的线性关系,取符合线性关系的探针浓度。另外,取偏振值对探针浓度作图,不同浓度的荧光探针在蛋白缓冲液中的偏振值理论上基本一致。取探针偏振值趋于稳定时的探针浓度。一般为了节省样品(探针浓度越高,到达饱和结合所需的蛋白就越多)且获得稳定的实验数据,取平行偏振光强度在1000~2000单位,偏振值稳定时对应的探针浓度。
例子:原始数据:
探针浓度(nM) | 500 | 250 | 125 | 62.5 | 31.25 | 15.625 | 7.8125 | 3.90625 | 1.953125 | 0 |
平行偏振光强度 | 7670 | 3967 | 2024 | 1168 | 731 | 307 | 173 | 147 | 62 | 24 |
偏振值 | 62 | 75 | 84 | 93 | 98 | 108 | 146 | 176 | 204 | 412 |
 |
6. 可选。蛋白偏振背景的检测。如果蛋白中添加有一些天然化合物建议做背景检测。
7. 结合常数测定。用稀释后的探针溶液将目的蛋白做浓度减半的系列稀释(此处蛋白最大浓度为150 Μm,体积为700 μL,之后取350 μL蛋白与相同体积探针混合并以此类推等体积减半稀释蛋白几次)。如果有沉淀,离心。
8. 在96孔板中每孔加入100 μL蛋白-探针混合液,每个浓度做3孔重复,包括一组不含蛋白的纯探针对照。96孔板离心去除气泡,1000 g×5-10 min。室温(温度根据需要选择)孵育一段时间,至偏振值稳定。孵育时间需要摸索,预实验可设定每30 min检测一次,一般如室温2小时。
9. 测量FP荧光信号(根据荧光分子选择合适的激发光波长及发射光波长)。具体操作根据软件而定。此处FAM荧光用BioTekR酶标仪配带的绿色偏振片(激发光485 nm和发射光528 nm)室温下进行偏振检测。
10. 数据处理。将测得的荧光偏振数据减去纯探针的背景信号得实际偏振值。将数据根据公式用GraphPad Prism软件进行非线性拟合分析,对蛋白浓度(X)-偏振值(Y)作图。根据实验结果调整蛋白浓度,如过早进入平台期则稀释蛋白浓度。(具体操作见附)
11. 普遍使用的荧光偏振拟合方程:

其中,B表示bing,LT指总配体浓度,RT指总受体浓度,Kd指结合常数。在实验中,配体或受体浓度可以一个保持恒定,改变另一个。
另外,另一个我们在中文文献中发现常被使用的荧光偏振拟合方程为Y=FPmin+FPmax*X/(X+kd)。
例子(这里我们使用后一方程进行数据拟合,得Kd=9.943 μM,R squared=0.9866)。

▲关键步骤:
♫ 荧光长时间曝光会发生荧光信号减弱现象,在实验过程中应避免过度曝光
♫ 单链DNA互补配对要缓慢退火,不能加热后直接放冰上
附:
1. FP实验中样品基本信息及实验原始数据
探针信息:探针序列为6-FAM-XXXXXXXXXXXXXXXXXX,探针互补序列为XXXXXXXXXXXXXXXXXX (X指某种碱基)。
探针稀释buffer为1×FP Buffer:5% glycerol, 80 mM NaCl,2 mM MgCl2,20 mM Tris-HCl pH 8.0
探针浓度: 62.5 nM
蛋白信息:蛋白P,分子量大于50 kD,自身不含核酸,室温下稳定。
实验原始数据 | ||||||||||
蛋白浓度(μM) | 150 | 75 | 37.5 | 18.75 | 9.375 | 4.6875 | 2.3437 | 1.17187 | 0.58593 | 0 |
偏振信号(mP)A | 108 | 109 | 105 | 98 | 89 | 79 | 74 | 68 | 70 | 67 |
偏振信号(mP)B | 110 | 108 | 103 | 96 | 87 | 83 | 75 | 73 | 64 | 66 |
偏振信号(mP)C | 109 | 107 | 104 | 97 | 89 | 79 | 76 | 71 | 67 | 64 |
每个实验组的实际偏振值=实验组偏振值-空白对照组(蛋白浓度为零)偏振值。再用实验组实际偏振值进行数据拟合。
2. 打开软件GraphPad Prism 8,选择左侧“New table & graph”下的“XY”,再选择右侧“Options”下方的“Y”,在第二行“Enter”前单击打钩,并在“Enter”后输入数字“3”,表示有三组平行数据。选择“Create”。


3. 填入实验数据,一列X(蛋白浓度)对应三组Y值(实际偏振值)。在数据页面选择数据上方的“Analyze”接着展开“XY analyses”并选择“Nonlinear regression (curve fit)”,在右上方“Analyze which data sets”下方选中要分析的数据后单击右下方“OK”。
4.在“Model”中选择并展开“binding saturation”,选择“one site-specfic binding”→OK。生成的“Table of results”中列出了数据拟合的具体信息,如下所示。R squaresd可以反映数据拟合的质量,越接近1表示越好。

▲ 关键步骤:选中“one site-specfic binding”后可以通过右方的“Details”查看拟合方程。方程选择的依据是具体的实验本身。如果多个结合位点及非特异性结合不能忽略时则不能使用该方程。一些文章中,蛋白-核酸亲和的荧光偏振实验所用方程为B={(LT + Kd + RT)-[(LT + Kd + RT)2- 4*LT*RT]^1/2}/2。
针对性建议(TROUBLESHOOTING)
因FP实验较为灵敏,建议在每次加样品之前检测96孔板的背景偏振值,选择各孔之间差异较小的孔进行实验。
3.4.7 等温滴定微量热Isothermal Titration Calorimetry
简介(INTRODUCTION)
等温滴定微量热(Isothermal Titration Calorimetry,ITC)是一种研究生物热力学与生物动力学的重要方法,它通过高灵敏度、高自动化的微量量热仪连续、准确地监测和记录一个变化过程的量热曲线,原位、在线和无损伤地同时提供热力学和动力学信息。通俗的讲就是将一种反应物滴到另一种反应物中,如果两者之间有相互作用,就会产热或吸热,热量的变化被检测到后,仪器会保持反应体系的温度与对照组的相同从而测得该过程中热量的变化(注意是保持温度不变测量热量变化,而不是直接测量温度变化)热量的变化与时间、浓度整合在一起,产生一个反应Kcal/mol、ligand/sample摩尔比等的滴定曲线。
微量热法操作简单(整个实验由计算机控制,使用者只需输入实验的参数,如温度、注射次数、注射量等,计算机就可以完成整个实验,再由Origin软件分析ITC得到的数据)。
ITC的用途:
获得生物分子相互作用的完整热力学参数,包括结合常数、结合位点数、摩尔结合焓、摩尔结合熵、摩尔恒压热容,和动力学参数(如酶活力、酶促反应米氏常数和酶转换数)。
ITC的应用范围:
蛋白质-蛋白质相互作用(包括抗原-抗体相互作用和分子伴侣-底物相互作用);蛋白质折叠/去折叠;蛋白质-小分子相互作用以及酶-抑制剂相互作用;酶促反应动力学;药物-DNA/RNA相互作用;RNA折叠;蛋白质-核酸相互作用;核酸-小分子相互作用;核酸-核酸相互作用;生物分子-细胞相互作用。
(横坐标为时间,纵坐标为热功率,一次每个峰面积为每次注射引起的热量总变化)
通过滴定操作和热量的测量,量热仪可以给出热量-摩尔比曲线:
图像中曲线的突跃中点对应的化学计量比就是两种蛋白质相互作用的化学计量数N,突跃中点处曲线的斜率就是两种蛋白质相互作用的结合常数Kd。
|
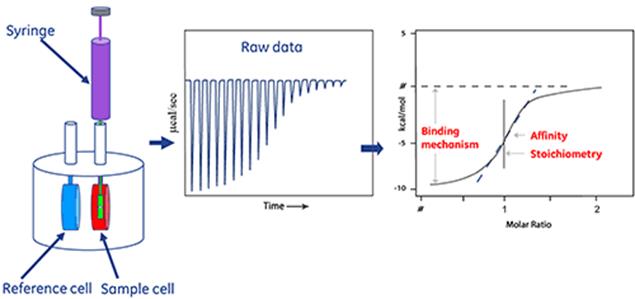
|

决定曲线形状的主操参数是C值:C=滴定池中的蛋白浓度/Kd。C值越大,曲线越陡;C值越小,曲线越平缓。一般C值在10-100之间实验效果最好。
材料(MATERIALS)
r 试剂(REAGENTS)
样品、buffer
r 器械(EQUIPMENT)
1 mL注射器若干,PCR管若干,1.5 mL EP管若干。
实验步骤(PROCEDURE)
1. 样品准备
1) 保证滴定物与被滴定物间的缓冲液保持完全一致
A. pH,盐浓度,DMSO浓度。
B. 同一缓冲液中透析或者置换大分子和配体。
C.若配体太小无法透析,用蛋白透析液配置(eg.小分子或者多肽)。
2)准确测量浓度
用A280准确测量蛋白浓度。尽量准确地称取配体。
3) 适用于广泛的缓冲体系
如需使用还原剂,最好使用β-巯基乙醇(β- mercaptoethanol,BME),而不能使用DTT。
4) 在样品准备完毕之后,要仔细测量pH,如果两者差值大于0.05,则其中的一个要重新用HCl或NaOH滴定到两者相差小于0.05。同时任何一种溶液都不能有沉淀,因此要过滤。
5) 两种溶液的最终溶度一定要精准,一次最好在样品完全准备好之后再次确定浓度(样品浓度对测得的参数影响很大)。
▲关键步骤:
♫ 基于ITC的原理,两种反应溶液的buffer完全相同就显得尤为重要。
♫ 还原剂的存在可能会引起基线的漂移,如果蛋白易沉淀,可加巯基乙醇。
2. 清洗机器
1)样品池:20% SDS、ddH2O、buffer
2)上样针:ddH2O、buffer
3.实验参数设定

4.滴定
1) 加样体积:
cell:300 μL,syringe:100 μL
测量Kd范围102~1012 M-1
2)滴定实验前恒温30~60 min
等温滴定量热实验所需时间,一般1.5-4 h
A. 实验温度设置一般为比室温,如果蛋白室温不稳定可低温。
B.一般第一次滴定0.4 μL,之后2 μL×19次,每次相隔120 s,第一次滴定的数据处理时通常丢弃。
C.对每个峰的面积进行积分,并以配体与蛋白质的摩尔比作为横坐标进行绘图。由此得出的等温线可拟合至导出亲和力(Kd)的结合模型。结合等温线中心的摩尔比即为反应化学计量。
5.数据处理
1) 读取数据
打开软件MicroCal LLC ITC200,点击Read data,找到自己的ITC数据,双击。

2) 检查浓度输入正确
点击左侧Data Control里的Concentration确认数据正确。

3) 调整Y坐标
点击上侧Math栏的Y Translate,双击图中的最后一个点平移到Y坐标0附近。

4)移除不好的数据
点击左侧Data Control里的Remove Bad Data,双击图中需要删除的点。

5) 拟合数据
点击Model Fitting中的One Set of Sites,点击100 lter两次,点击1 lter多次至Reduced Chi-sqr数字基本不变,点击Done。
6)作图
将出现的数据图copy到上侧ITC栏的Final Figure页面。


7)保存图
点击上侧File栏的Export Page,选择需要的图片类型,保存到合适文件夹。
针对性建议(TROUBLESHOOTING)
1. 如果对照组每次滴定引起的热量变化都很小且大小平均的化,在处理数据时可以从样品组中直接减去。
2. 如果对照组的热量变化也比较大,可能是两种Buffer不匹配,如果buffer确实是匹配的化,可能是由于配基在syringe中发生了聚合。
3. 原始的数据是横坐标为时间,纵坐标为热量变化的图,处理后为横坐标为摩尔比,纵坐标为Kcal/mol。
3.4.8 表面等离子共振技术Biacore
简介(INTRODUCTION)
表面等离子共振(SPR)是一种光学现象,可被用来实时跟踪在天然状态下生物分子间的相互作用。这种方法对生物分子无任何损伤,且不需任何标记物。光在棱镜与金属膜表面上发生全反射现象时,会形成消逝波进入到光疏介质中,而在介质(假设为金属介质)中又存在一定的等离子波。当两波相遇时可能会发生共振。当消逝波与表面等离子波发生共振时,检测到的反射光强会大幅度地减弱。能量从光子转移到表面等离子,入射光的大部分能量被表面等离子波吸收,使反射光的能量急剧减少,这就是SPR的光学原理。
表面等离子共振广泛应用于研究结合特异性、抗体选择、抗体质控、疾病机制、药物发明、生物治疗、生物处理、生物标记物、配体垂钓、基因调控、细胞信号传导、亲和层析、结构-功能关系、小分子间相互作用等。
表面等离子共振(SPR)是一种光学现象,可被用来实时跟踪在天然状态下生物分子间的相互作用。这种方法对生物分子无任何损伤,且不需任何标记物。
先将一种生物分子(靶分子)键合在生物传感器表面,再将含有另一种能与靶分子产生相互作用的生物分子(分析物)的溶液注入并流经生物传感器表面。生物分子间的结合引起生物传感器表面质量的增加,导致折射指数按同样的比例增强,生物分子间反应的变化即被观察到。这种反应用反应单位(RU)来衡量:1 RU=1 pg蛋白/mm2 =1x10-6 RIU(折射指数单位)。

分析物在被注入的过程中,由对流和扩散流经相互作用表面而与靶分子形成复合物,导致分析物浓度改变。微射流系统内nL数量级流动通道的应用,使得这种浓度的改变降至最低点,以确保高传质系数(Mass Transport Coefficient,km)。为保证分析物的传质性不被限制,键合在生物传感器表面的靶分子浓度必须较低。当分析物被注入时,分析物-靶分子复合物在生物传感器表面形成,导致反应增强。而当分析物被注入完毕后,分析物-靶分子复合物解离,导致反应减弱。通过结合式相互作用模型拟合这种反应曲线,动力学常数便可被确定。而非特异性结合和总折射指数移相等效应则可通过参照曲线减除功能予以驱除。
在操作过程中,任何一对亲和分子,一个(靶分子)被固定在生物传感器(芯片)表面,另一个(分析物)被置于溶液中。当含有分析物的溶液流经靶分子固定的生物传感器表面时,亲和性复合物形成。
如果要得到亲和力数据,学院实验中心有:没有标签的蛋白一般用CM5芯片,带biotin的用SA,NTA一般用于定性分析。一张芯片有4个通道可以偶联2个不同样品。
材料(MATERIALS)
r 试剂(REAGENTS)
如若使用氨基偶联的方法,靶分子不能置于含有Tris的buffer中,可以使用HEPES buffer。分析物可以使用Tris buffer。
0.22 μm滤膜过滤的新鲜的Hepes buffer及Tris buffer。
靶分子需要1 mg/mL或者以上,几十微升。
分析物需要1 mg/mL或者以上,定量分析需要几百微升。
几个EP管
实验步骤(PROCEDURE)
1. 以氨基偶联为例,偶联于芯片上的蛋白通过氨基随机偶联,故蛋白不能处于Tris 环境中,在制备蛋白样本时,应将buffer置换成无Tris的溶液如HEPES buffer。而作为流动相的蛋白分子不需要将buffer置换。
2. 进行实验的当天,用0.22 μm滤膜抽滤两种蛋白所在的buffer即Tris buffer 与HEPES buffer,各400 mL。
3. 带上所有物品去生院三楼仪器中心进行试验。
4. 先将芯片取出,放入机器,使用非Tris的buffer prime 两遍(置换系统中的buffer)。
5. 每张芯片存在四个通道,一次使用两个通道,一个为空白对照通道,一个偶联蛋白。确定哪个通道为对照,哪个为偶联蛋白的实验通道。
6. 上样偶联时确定最大不超过150个RU,通过蛋白质PI,MW,浓度等确定上样量为多少微升。
7. 偶联后使用Tris buffer封闭多余位点。
8. 再将系统置换buffer为Tris buffer,prime两遍。
9. 确定流动相上样量,进行试验。
10. 实验结果分析:

该图展示为CARD 固定于芯片上,LPS 为流动相,4通道为实验通道,3通道为对照通道。故当不同浓度的LPS依次流穿3,4通道后,会形成两个不同的response曲线。而确定是否存在相互作用,可以通过“实验通道-对照通道”来确定是否存在相互作用。一般几个RU的反应就可以看到曲线明显的变化。但是这里展示的就没有变化,即CARD 与LPS 无相互作用。
3.4.9 蛋白质交联Crosslink
简介(INTRODUCTION)
 |
Crosslink是一个很常用的检测蛋白相互作用的技术,用来检测蛋白质在溶液中的互作。它有很多不同种类的交联剂(crosslinker),本实验室用的linker是DSS,以下是其结构。DSS是一个水溶的homobifunctional N-hydroxysuccinimide ester(简称NHS-ester)。
当两个蛋白有相互作用时,其距离会很近,NHS-ester可以与蛋白的primary amines(比如赖氨酸的残基末端)反应从而在两个蛋白之间形成共价键后的两个蛋白在SDS环境下是不会分开的,因此我们可以通过梯度变性胶来检测复合物的存在与否。
那么DSS的空间长度(11.4 Å)是否能满足共价键的形成呢?下面是有机体中主要的几个共价键长度。
有机体中共价键长度表

DSS与蛋白形成共价键是以C-H形式形成的,因此其空间长度11.4 Å足以作为桥梁。但需要注意的是DSS通过primary amine与蛋白质形成共价键,因此不能使用像Tris等含有amine的溶液,需要把蛋白透析到含有HEPES的缓冲液中。
Crosslink不仅可以在溶液中检测两个蛋白的相互作用,也可以检测在细胞内甚至体内中某几个蛋白的相互作用。在此我们主要介绍溶液中两个纯化好的蛋白之间的crosslink方法。
材料(MATERIALS)
r 试剂(REAGENTS)
DMSO、Conjugation buffer:Use a non-amine-containing buffer with a pH 7~9、Quenching buffer(终止液):1 M Tris-HCl pH 7.5(或者1 M glycine 1 M lysine solution 也可以)
r 实验前准备(SETUP)
将蛋白质所处的溶液换成Conjugation buffer
r 器械(EQUIPMENT)
蛋白质电泳仪、Scan-drop、照胶仪、金属浴
实验步骤(PROCEDURE)
1. 摇菌 ►预计时间消耗:2~4小时
取2 mg DSS用DMSO或者DMF溶解到需要的浓度,如下表:
Solvent volume to add to 2 mg DSS (μL) | Buffer volume to add to 2 mg BS3 (μL) | Crosslinker Conc. (mM) |
432 | 277 | 12.5 |
216 | 140 | 25 |
108 | 70 | 50 |
54 | 35 | 100 |
2. 准备样品。需要先测量样品的浓度,根据其分子量和需要的体积来计算摩尔。如果浓度大于5 mg/mL,需要10倍摩尔的DSS;如果浓度小于5 mg/mL,需要20~50倍摩尔的DSS。DSS终浓度需要控制在0.25~5 mM。
3.混匀后在室温放置半小时,或者在冰上放置2小时。
4. 加入终止液,使得终止液的终浓度在20~50 mM范围内。直接加入SDS-loading buffer即可,因为其中已经含有Tris。
5. 室温放置15分钟,在100 °C金属浴中加热10分钟,跑梯度胶。
▲关键步骤:溶液中的蛋白质要很纯,不然跑胶后的条带会很乱。
现以TNFR1和DR3为例,按照上述步骤进行实验,得到下图:

Crosslink 结果图:
蛋白浓度:2 mg/mL 5、6:阴性对照组
由上图可见,crosslink实验必须选好阴性对照,本例中阴性对照也有部分聚合,实验组聚合更加明显。所以crosslink更适合用于粗略的定性,作为辅助性结果,想要确证蛋白是否聚合还需要其他更有说服力的实验作为支持。
针对性建议(TROUBLESHOOTING)
1. 带tag的蛋白,如MBP conjugated protein,需要用MBP来做阴性对照。
2. 即使不带任何tag,也需要设置阴性对照和阳性对照。阴性对照可以使MBP这种已知单体的蛋白,阳性对照可以选择已经证明有相互作用的蛋白。从而保证实验系统本身的可行性。
3.4.10 Native-PAGE
简介(INTRODUCTION)
Native-PAGE(非变性聚丙烯酰胺凝胶)依据蛋白质所带电荷、分子量及形状的差异分离折叠的蛋白、蛋白-蛋白或蛋白-配体复合物。
Native-PAGE和变性SDS-PAGE电泳在操作上基本相同,只是Native-PAGE要求蛋白处在非还原、非变性环境中以维持蛋白的二级结构及天然的电荷密度。所以蛋白样品、凝胶及电泳缓冲液中均不能含有变性剂如SDS 等。进行非变性凝胶电泳前要依据蛋白等电点(PI)来分清靶蛋白是碱性还是酸性蛋白。分离酸性蛋白,用高pH凝胶系统,分离碱性蛋白,用低pH凝胶系统。酸性蛋白或微碱性蛋白(pI = 3~8)采用pH 8.3的缓冲系统,蛋白带负电荷,向阳极移动;而碱性蛋白(pI > 8,9)在微酸性环境中进行,蛋白带正电荷,将阴极和阳极倒置进行电泳。
用途:
检测蛋白间及蛋白与配体之间的相互作用
检测蛋白同种型或构象异构体
|

材料(MATERIALS)
r 试剂(REAGENTS)
分离酸性蛋白 | 分离碱性蛋白 |
30%丙烯酰胺 | 同左 |
(A液:0.5 M Tris-HCl pH 6.8, B液:1.5 M Tris-HCl pH 8.8)或5×TBE | A液:0.25 M Acetate-KOH pH 6.8, B液:1.5 M Acetate-KOH pH 4.3 |
10% APS | 同左 |
ddH2O | 同左 |
TEMED | 同左 |
溴酚蓝-loading buffer(2×) | 甲基绿-loading buffer(5×) |
电泳Buffer:0.5×TBE或者pH 8.3 Tris-Gly | 电泳Buffer:pH 4.5 β-丙氨酸-Ac |
r 器械(EQUIPMENT)
制胶架、电泳仪、电泳槽、离心机
实验步骤(PROCEDURE)
分离酸性蛋白
1. 配制工作液
1)30%丙烯酰胺、10% APS详见SOP SDS-PAGE部分
2)A液:0.5 M Tris-HCl pH 6.8:6 g Tris base 溶于80 mL水,浓HCl调pH 6.8,加水定容到100 mL。室温贮存
3) B液:1.5 M Tris-HCl pH 8.8:18.2 g Tris base溶于80 mL水,浓HCl调pH 8.8,水定容到100 mL。室温贮存
4) 10×电泳Buffer:
5) pH 8.3 Tris-Gly:30.3 g Tris base,144.1 g甘氨酸,加水定容到1 L,不用调pH。室温贮存
6)或者5× TBE:27 g Tris base,13.75 g硼酸,10 mL 0.5 M EDTA pH 8.0,定容到500 mL。室温贮存
7) 溴酚蓝-loading buffer(2×):125 μL 0.5 M Tris-HCl pH 6.8,360 μL甘油,0.5 mg溴酚蓝,加dH2O至1 mL。(62.5 mM Tris-HCl pH 6.8,25~36% glycerol,1% Bromophenol Blue)-20 °C贮存
2. 凝胶制备
1) 气密性检测:装配凝胶架,并用自来水检测凝胶槽气密性,无漏液为佳。倒掉胶板之间的水并用滤纸吸干备用。
2) 配胶:本实验室1.0 mm规格的制胶板可容纳6 mL凝胶/块,故配方以制备一块凝胶的量为例。
单层非变性胶(仅分离胶)配方(12%)
总体积 | 10 mL | 6 mL |
ddH2O | To 10 mL | To 6 mL |
30% 丙烯酰胺 | 4 mL | 2.4 mL |
5×TBE或者B液 | 2 mL或B液2.5 mL | 1.2 mL或B液1.5 mL |
10% AP | 60 μL | 36 μL |
TEMED | 15 μL | 8 μL |
双层非变性胶配方(12%)
12%分离胶 10 mL | 5% 浓缩胶 4 mL |
30%丙烯酰胺 4 mL | 30%丙烯酰胺 670 μL |
1.5 M Tris-HCl pH 8.0 2.5 mL | 0.5 M Tris-HCl pH 6.8 1 mL |
10% APS 50 μL | 10% APS 30 μL |
ddH2O 3.445 mL | ddH2O 3.3 mL |
TEMED 5 μL | TEMED 5 μL |
A. 灌胶:10% APS和TEMED在灌胶前加入,轻轻摇匀并立即使用。用1 mL移液器或吸管吸取胶液并从注胶板内侧的一角将其灌注到注胶板之间。制备单层胶,注满后立即加梳子。而制备双层胶,先加注分离胶液面到距离胶板顶部0.8~1 cm,用1 mL ddH2O或者异丙醇封闭,静置约30 min待胶凝固后倒掉并吸干胶面水或异丙醇,配置浓缩胶灌满胶板,加梳子静置30~60 min至胶体凝固。
B. 保存:凝胶和玻璃板一起置于7%乙酸溶液中密封储存2~3个月。
浓度与体积换算公式:
×% 分离胶 10 mL |
30%丙烯酰胺 ×/3 mL (终浓度1%) |
1.5 M Tris-HCl pH 8.8 2.5 mL |
10% APS 50 μL |
ddH2O (7.5- ×/3)mL |
TEMED 5 μL (若×< 8%,取10 μL) |
3. 样品制备
取蛋白样加入等体积的loading buffer(2×)混匀,不要加热,10 000 g×5 min,取上清上样(载样过多会使电泳条带变形)。
4. 电泳
预电泳(加样前电泳):0.5×TBE 电泳buffer (编者常用)或者pH 8.3 Tris-Glycine buffer,冰浴电泳30-60 min。再上样,电泳约3小时。电泳电压不低于100 V,不高于200 V。电压结束后回收电泳buffer重复使用。
▲关键步骤:电泳buffer 应在pH 8.3附近。不调pH,如果pH不在此范围,要弃去重新配制。
5. 脱染色
按照常规SDS-PAGE胶方法进行染色脱色,“清水煮沸2 minà G250煮沸2 minà清水煮沸2 min”,直至胶板背景透明。也可以用银染或者活性染色。也可以不进行染色处理,直接采集图像。
染色方法 | 灵敏度 |
Coomassie blue (G-250) | 0.2~0.5 μg/band |
Silver staining | 2 ng/band |
Fluorescent dye staining | 5 ng/band |
分离碱性蛋白
1. 配制工作液
1) A液:0.25 M Acetate-KOH pH 6.8:48 mL 1 M KOH,2.9 mL HAc,加ddH2O至200 mL。室温贮存
2) B液:1.5 M Acetate-KOH pH 4.3:48 mL 1 M KOH,17.2 mL HAc,加ddH2O至200 mL。室温贮存
3) 甲基绿-loading buffer(5×):333 μL 0.25 M Acetate-KOH pH 6.8,483 μL甘油,0.5 mg甲基绿,dH2O 172 μL至终体积为1 mL。-20 °C贮存。
4) 10×电泳Buffer:18.7 g β-丙氨酸,4.8 mL HAc,加水到600 mL,调pH 4.5(0.35 M β-丙氨酸,0.14 M HAc pH 4.5)。室温贮存。
2. 凝胶制备
同分离酸性蛋白相同,只是改用酸性系统的A/B液。
12% 分离胶 10 mL | 5% 浓缩胶 4 mL |
30%丙烯酰胺 4 mL | 30%丙烯酰胺 667 μL |
1.5 M Acetate-KOH pH 4.3 2.5 mL | 0.25 M Acetate-KOH pH 6.8 1 mL |
10% APS 50 μL | 10% APS 30 μL |
ddH2O 3420 μL | ddH2O 3300 μL |
TEMED 30 μL | TEMED 5 μL |
▲关键步骤:
pH值低于6时,TEMED的催化效率会降低。需增加5倍的TEMED浓度在pH 4-6的凝胶中使之聚合。催化剂的溶度对于凝胶聚合非常重要!厚边现象和胶孔形状不完整往往由它引起。
3. 样品制备。取蛋白样加入1/4样品体积的甲基绿-loading buffer(5×)混匀,不加热,10 000 g×5 min,取上清上样。
4. 电泳。预电泳(加样前电泳):β-丙氨酸-Ac电泳buffer,将正负电极倒置,冰浴电泳30~60 min。再上样,电泳约3小时。电泳电压不低于100 V,不高于200 V。电压结束后回收电泳buffer重复使用。
▲ 关键步骤:正负电极倒置不可少,脱染色的实验操作同酸性蛋白的分离。
针对性建议(TROUBLESHOOTING)
由于此过程中选用了几个pH环境,要根据蛋白自身的特性选择稳定蛋白的pH环境。如果该缓冲体系不适合(如带电性质导致蛋白不发生电泳迁移,蛋白沉淀,缓冲体系与检测系统不兼容),可选用其它缓冲体系,具体情况请参阅相关资料。
日常维护:
缓冲液槽及上盖,电极芯、共用组件、制胶架及框 | 使用后用蒸馏水彻底冲洗
|
玻璃板及电泳梳子 | 清洁剂清洗后用蒸馏水彻底冲洗干净。带边条玻璃板在强碱溶液,如>100 mM NaOH,中浸泡不能超过24小时;在玻璃洗液中浸泡不能超过2小时。长时间的浸泡将会破坏边条粘合的完整性。为延长边条粘合的寿命,须避免在碱性清洗液中浸泡超过5天 |

常见问题与解决方案(COMMON PROBLEMS AND SOLUTIONS)
|

3.4.11 静态光散射Static light scattering
简介(INTRODUCTION)
静态光散射(SLS)是一项测量绝对分子量的技术,如瑞利理论所述,该技术利用一个分子散射的光强度与其分子量和尺寸之间的关系进行测量。简言之,瑞利理论是指,特定光源条件下,较大分子与较小分子相比会散射更多的光,而散射的光强度与分子的分子量互成比例。通过测量多角度下的散射强度去计算均方根半径,即回旋半径Rg。
材料(MATERIALS)
r 试剂(REAGENTS)
超声或高压过的ddH2O
超声或高压过的buffer
总量大于等于1 mg的样品(根据使用的loop决定体积大小)
r 器械(EQUIPMENT)
500 μL或1 mL的上样环
1mL注射器
事先平衡的24 mL分子筛
实验步骤(PROCEDURE)
清洗仪器管路及平衡
1. 仪器系统中充满的是20%乙醇,故先用高压或超声过后无气泡的ddH2O以0.2 mL/min的流速开始清洗系统,当压力稳定后,可适当调高流速,一共清洗大概25 mL即可。
2. 使用buffer再次清洗系统,步骤依旧是从低流速开始,压力稳定后适当调高流速,清洗25 mL。
3. 连接柱子到纯化仪上,根据柱子压力以及要求确定流速,连接后,平衡2~4 h即可,观察光散射仪上的基线时候波动不剧烈,LS曲线在10-4以下即可。
上样检测
1. 根据使用说明将ASTAR7软件打开选择好实验,并在该选项中更改流速为柱子限定流速 0.6 mL/min。
2. 其后在procedures下选择basic collection,根据流速确定duration(如0.6 mL/min的流速,24 mL的柱子,则duration(min)设置为40 min,可确定跑完24 mL的柱子)。


3. 完成设置后,在ASTAR7界面中点击菜单栏的run按钮 ,等待一个窗口弹出,询问是否开始实验,先不点击确定,此时上样于上样环中,在AKTA界面切换Mannal load为Inject后,切换到ASTAR7界面在弹窗中点击确定,即两边仪器同时开始工作。
,等待一个窗口弹出,询问是否开始实验,先不点击确定,此时上样于上样环中,在AKTA界面切换Mannal load为Inject后,切换到ASTAR7界面在弹窗中点击确定,即两边仪器同时开始工作。
4. 在实验完成后,卸下上样环,恢复仪器原样,按照仪器说明书,需要用高压或超声的ddH2O清洗系统4个小时。
5. 其后将系统中的溶液置换成20%乙醇。
数据处理
1. 在“Procedures”中选择“Baselines”,选择LS5,确定基线(baseline)以后,选择“set all”,再选择UV确定基线(baseline)。将基线即为无样品时基准对照值。两条线分别确定基线,最后点击apply。

2. 其后选择“Peaks”,确定你所计算分子量的数据取哪些,如果样品存在两个峰,在第二个峰的开始位置点击鼠标左键即创建另外一个peak,选择好后点击apply,同时需要更改蛋白样品的吸光系数,这直接影响后续软件的计算结果。

3. 在“Results fitting”中确定数据选取的质量,MW的曲线应该较为平稳,上下浮动不明显。

4. 最后于“Results”中选择“Report(summary)”确定MW以及误差范围。

5. 数据导出,在“Peaks”界面图上单击右键,选择“Edit”,选择“Export”,点击确定,导出对应peak的MW数据。而UV数据可以通过选择菜单栏中的“File”“Export”,保存类型选择CSV导出数据,两个数据可以通过excel打开,再作图。

3.4.12 凝胶阻滞实验EMSA-electrophoretic mobility shift assay
简介(INTRODUCTION)
电泳迁移率变动分析(EMSA-electrophoretic mobility shift assay),也称凝胶阻滞分析,是用于研究蛋白质-DNA或蛋白质-RNA相互作用的常用技术。
通常将纯化的蛋白和细胞粗提液和32P同位素标记的DNA或RNA探针一同保温,在非变性的聚丙烯凝胶电泳上,分离复合物和非结合的探针。DNA-复合物或RNA-复合物比非结合的探针移动得慢。同位素标记的探针依研究的结合蛋白的不同,可是双链或者是单链。当检测如转录调控因子一类的DNA结合蛋白,可用纯化蛋白、部分纯化蛋白或核细胞抽提液。在检测RNA结合蛋白时,依据目的RNA结合蛋白的位置,可用纯化或部分纯化的蛋白,也可用核或胞质细胞抽提液。竞争实验中采用含蛋白结合序列的DNA或RNA片段和寡核苷酸片段(特异),和其它非相关的片段(非特异),来确定DNA或RNA结合蛋白的特异性。在竞争的特异和非特异片段的存在下,依据复合物的特点和强度来确定特异结合。
材料(MATERIALS)
r 试剂(REAGENTS)
DTT、BSA、10×TBE buffer TBE-PAGE gel、[γ-32P]ATP、T4多聚核苷酸激酶、Nuclease-Free Water、T4多聚核苷酸激酶缓冲液、醋酸铵、TE、无水乙醇
EMSA/Gel-Shift结合缓冲液(5×):50 mM Tris HCl pH 8.0,750 mM KCl,2.5 mM EDTA,0.5% Triton-X 100,62.5% glycerol (v/v),1 mM DTT add DTT fresh before use
r 器械(EQUIPMENT)
PAGE胶电泳设备
实验步骤(PROCEDURE)
1.探针的标记
1)体系建立
反应体系(10 μL) |
|
待标记探针 (1.75 pmol/μL) | 2 μL |
T4 Polynucleotide Kinase Buffer (10×) | 1 μL |
Nuclease-Free Water | 5 μL |
[γ-32P]ATP(3000 Ci/mmol at 10 mCi/mL) | 1 μL |
T4 Polynucleotide Kinase (5~10 U/μL) | 1 μL |
按照上述反应体系依次加入各种试剂,加入同位素后,Vortex混匀,再加入T4 Polynucleotide Kinase,混匀。
▲ T4多聚核苷酸激酶的用途:
DNA或RNA的末端标记,用作探针和进行DNA测序
DNA或RNA 5’末端的磷酸化,以便进行连接反应
移除3’端磷酸集团
2) 使用水浴或PCR仪,37 °C反应10分钟。
3)加入1 μL探针标记终止液,混匀,终止探针标记反应。
4)再加入89 μL TE,混匀。此时可以取少量探针用于检测标记的效率。通常标记的效率在30%以上,即总放射性的30%以上标记到了探针上。为实验简便起见,通常不必测定探针的标记效率。
▲ 标记好的探针最好立即使用,最长使用时间一般不宜超过3天。标记好的探针可以保存在-20 °C。
2 探针的纯化
通常为实验简便起见,可以不必纯化标记好的探针。在有些时候,纯化后的探针会改善EMSA的电泳结果。如需纯化,可以按照如下步骤操作:
1) 对于100 μL标记好的探针,加入1/4体积即25 μL的5 M醋酸铵,再加入2体积即200 μL的无水乙醇,混匀。
2) 在-70 °C至-80 °C沉淀1小时,或在-20 °C沉淀过夜。
3) 在4 °C,12000 g~16000 g离心30分钟。小心去除上清,切不可触及沉。
4) 在4 °C,12000 g~16000 g离心1分钟。小心吸去残余液体。微晾干沉淀,但不宜过分干燥。
5) 加入100 μL TE,完全溶解沉淀。标记好的探针最好立即使用,最长使用时间一般不宜超过3天。标记好的探针可以保存在-20 °C。
3 EMSA 胶的配制(注意:使用29:1等不同比例的Acr/Bis对结果影响不大)。
4%的聚丙烯酰胺凝胶 | 20 mL |
TBE buffer (10×) | 1 mL |
ddH2O | 16.2 mL |
39:1 acrylamide/bisacrylamide (40%,w/v) | 2 mL |
80% 甘油 | 625 μL |
10% 过硫酸铵 (ammonium persulfate) | 150 μL |
TEMED | 10 μL |
4 EMSA结合反应
实验组体系 |
|
Nuclease-Free Water | 5 μL |
EMSA/Gel-Shift结合缓冲液(5×) | 2 μL |
100×BSA | 0.1 μL |
细胞核蛋白或纯化的转录因子 | 2 μL (5 μg protein total) |
标记好的探针 | 1 μL |
总体积 | 10 μL |
阴性对照 | 实验组 | 探针冷竞争反应 | 突变探针的冷竞争反应 | Super-shift反应 |
不加蛋白 | 加蛋白 | 比实验组多加未标记的探针1 μL | 比实验组多加未标记的突变探针1 μL | 比实验组多加目的蛋白特异抗体1 μL |
1) 按照上述顺序依次加入各种试剂,在加入标记好的探针前先混匀,并且室温(20~25 °C)放置10分钟,从而消除可能发生的探针和蛋白的非特异性结合,或者让冷探针优先反应。然后加入标记好的探针,混匀,室温(20~25 °C)放置20分钟。
2) 加入1 μLEMSA/Gel-Shift上样缓冲液(无色,10×),混匀后立即上样。注意:有些时候溴酚蓝会影响蛋白和DNA的结合,建议尽量使用无色的EMSA/Gel-Shift上样缓冲液。如果对于使用无色上样缓冲液在上样时感觉到无法上样,可以在无色上样缓冲液里面添加极少量的蓝色的上样缓冲液,至能观察到蓝颜色即可。
5 电泳分析
1) 用0.5×TBE作为电泳液。按照10V/厘米的电压预电泳10分钟。预电泳的时候如果有空余的上样孔,可以加入少量稀释好的1×的EMSA上样缓冲液(蓝色),以观察电压是否正常进行。
2) 把混合了上样缓冲液的样品加入到上样孔内。在多余的某个上样孔内加入10 μL稀释好的1×的EMSA/Gel-Shift上样缓冲液 (蓝色),用于观察电泳进行的情况。
3) 按照10 V/厘米的电压电泳。确保胶的温度不超过30 °C,如果温度升高,需要适当降低电压。电泳至EMSA/Gel-Shift上样缓冲液中的蓝色染料溴酚蓝至胶的下缘1/4处,停止电泳。
针对性建议(TROUBLESHOOTING)
1. 实验中需要用到什么试剂?凝胶迁移实验需要的结合蛋白,可来源于纯化或部分纯化的蛋白,或粗的核和胞质抽提液。还必须制备同位素标记的DNA或RNA。一般,DNA核苷酸探针用32P和T4多核苷酸激酶来作末端标记,同位素标记的RNA用噬菌体RNA聚合酶和同位素标记的核苷酸在体外合成。Promega公司的Riboprobe/sup系统(a,b)可用于同位素标记的RNA的体外合成,DNA 5’末端标记系统,用于制备DNA探针,结合反应所需的组分有:含盐的溶液(氯化镁,氯化钠,或氯化钾)、缓冲体系(Tris-HCl或HEPES)、还原剂(DTT)、甘油、非特异的竞争DNA(poly(dI:dC)dI:dC),也可能含非离子去污剂。在结合蛋白和同位素标记的探针作用后,在非变性的聚丙烯凝胶电泳上,分离复合物,随后将凝胶干燥并放射自显影,或用PhosphorImage/sup分析。
2. 成功进行凝胶迁移实验,需要优化哪些因素?凝胶迁移实验在理论上很简单也很快速,但要成功地进行凝胶迁移实验,需要优化一些参数,这主要受结合蛋白的来源和探针结合位点特点的影响。以下是需要优化的因素:抽提液的制备(核酸酶和磷酸酶污染会使探针降解),结合蛋白的浓度,探针的浓度,非特异性探针的浓度,缓冲液的配方和pH,聚丙烯凝胶电泳的特点和电泳条件,保温时间和温度,载体蛋白,是否有辅助因子(比如锌,或镉等金属离子,或激素)。总之,反应总体积应最小20 μL。为满足一般要求,结合缓冲液含4%甘油,1 mM MgCl2,0.5 mM EDTA,0.5 mM DTT,50 mM NaCl,10 mM Tris-HCl pH 7.5,0.05 mg/mL poly dI:dC,或10 mM HEPES pH 7.9,50 mM KCl,1 mM DTT,1 mM EDTA,10%甘油,0.05 mg/mL poly dI:dC可作为优化实验的起始。
3. 在DNA探针的选择上,要考虑哪些重要因素?
目的DNA的长度应小于300 bp,以有利于非结合探针和蛋白DNA复合物的电泳分离。双链的合成的寡核苷酸和限制性酶切片段可在凝胶迁移实验中用作探针。如目的蛋白已被鉴定,则应用短的寡核苷酸片段(约为25 bp),这样结合位点可和其他因子的结合位点区别开。长的限制性酶切片段可用于对推定的启动子/增强子区域内的蛋白结合位点定位。随后可用DNaseI 印迹对蛋白结合的特异区域在DNA序列水平上作出分析。
3.5 蛋白生化实验技术
3.5.1 蛋白酶活性检测
简介(INTRODUCTION)
蛋白酶是水解蛋白质肽链的一类酶的总称。按其降解多肽的方式分成内肽酶和端肽酶两类。内肽酶可把大分子量的多肽链从中间切断,形成分子量较小的肽;端肽酶又分为羧肽酶和氨肽酶,它们分别从多肽的游离羧基末端或游离氨基末端逐一将肽链水解生成氨基酸。
按其活性中心和最适pH值,又可将蛋白酶分为丝氨酸蛋白酶、巯基蛋白酶、金属蛋白酶和天冬氨酸蛋白酶。按其反应的最适pH值,分为酸性蛋白酶、中性蛋白酶和碱性蛋白酶。工业生产中应用的蛋白酶主要是内肽酶。
 这里以检测寨卡病毒NS2B-NS3丝氨酸蛋白酶活性为例,希望能对其他蛋白酶的活性分析提供参考。丝氨酸蛋白酶酶活分析的原理是它能特异性剪切人工合成的含有其识别位点的荧光多肽(EDANS—Cleavage site---Dabcyl)底物。
这里以检测寨卡病毒NS2B-NS3丝氨酸蛋白酶活性为例,希望能对其他蛋白酶的活性分析提供参考。丝氨酸蛋白酶酶活分析的原理是它能特异性剪切人工合成的含有其识别位点的荧光多肽(EDANS—Cleavage site---Dabcyl)底物。
如图所示,EDANS的激发光波长是336 nm,发射光波长是490 nm,而Dabcyl的激发光波长是472 nm。当底物未被剪切时,EDANS和Dabcyl距离较近,后者吸收前者发射的光而被激发,在490 nm处检查不到供体的荧光,而当底物被剪切时,供体和受体距离较远,光不能被受体吸收,可以检测到供体发出的490 nm的荧光。据此检查蛋白酶活性。
酶活力单位可以通过测定一定温度和pH值条件下,单位时间内底物消耗量或产物增加量来表示。由于该蛋白酶是一种米氏酶,所以这里以米氏常数(Km)来表征蛋白酶的活力特征。
材料(MATERIALS)
r 试剂(REAGENTS)
蛋白酶、荧光多肽底物、NaCl、甘油、去污剂CHAPS
缓冲能力pH 5~11的1 M缓冲液(MES pH 5.5~6.5、MOPS pH 6.5~7.5、Tris-HCl pH 7.5~9.0、CHES pH 9.0~10.0、CAPS pH 10~11.0)
二价金属离子的氯盐(10 mM 的MgCl2、CaCl2、ZnCl2、MnCl2)
gel filtration buffer:100 mM NaCl,10 mM Tris-HCl pH 8.0
基础酶切buffer:50 mM CHES pH 9.5,30% glycerol,1 mM CHAPS
r 器械(EQUIPMENT)
黑色96孔板、12孔排枪、离心机、酶标仪
实验步骤(PROCEDURE)
1. EDANS标准曲线的绘制
不加酶时,底物的EDANS的发射光被Dabcyl吸收,所以EDANS标准曲线需要通过检测不同浓度下EDANS试剂的发射光强度来绘制,纵坐标为荧光读数,横坐标为试剂浓度。
用水将EDNAS稀释至1000,500,250,125,62.5,31.25,15.6,7.8,3.9,0 nM。在黑色96孔板上加入不同浓度的EDNAS,用基础酶切buffer补至每孔100 μL,每孔3个重复。轻轻振板1 min,酶标仪室温检测336/490 nm波长的荧光强度,绘制标准曲线。
2. 蛋白酶的酶反应曲线
在黑色96孔板中加入基础酶切buffer、0.1 µM蛋白酶,离心混匀,设置酶标仪运行参数,再用排枪向96孔板中加入100 µM荧光底物。使每孔100 μL,每孔3个重复。轻轻振板1 min ,每隔1 min室温检测336/490 nm波长的荧光强度并绘制酶反应曲线。
 |
| ||

结果反应了蛋白酶在当前反应体系下的30 min内呈现一级反应的特征。可在一级反应时间内检测蛋白酶的其他特征。
► 最适底物浓度及蛋白浓度
酶浓度恒定,探究底物量
查阅相关文献,首先选定一个蛋白酶浓度区间进行实验。这里选了50 nM和200 nM蛋白酶作为恒定浓度,分别探究了其与底物浓度比例分别为1:200、1:400、1:800和1:500、1:1000的酶活变化。

在黑色96孔板中加入基础酶切buffer、蛋白酶,离心混匀,设置酶标仪运行参数,再用排枪向96孔板中加入相应量的荧光底物。使每孔100 μL,每孔3个重复。轻轻振板1 min,每隔0.5 min室温检测336/490 nm波长的荧光强度并绘制酶反应曲线。
结果分析:50 nM蛋白酶在底物未饱和情况下的水解速率和底物浓度呈正相关,而在200 nM蛋白酶时,底物的增加对蛋白酶反应速率的影响不大。所以认为,如果检测前10 min的蛋白酶消化水平,蛋白酶浓度不高于200 nM为宜。如果实验需要,还可以在50 nM的基础上适当降低蛋白酶浓度。
▲关键步骤:
♫ 蛋白酶用量不能过少,否则溶液中不稳定因素占据上风会使实验误差不容忽略。
♫ 底物浓度恒定,探究蛋白酶量
♫ 为节约底物的需要,探究底物浓度恒定时的蛋白酶用量是合理的。在黑色96孔板中加入基础酶切buffer和不同终浓度的蛋白酶,离心混匀,设置酶标仪运行参数,再用排枪向96孔板中加入5 μM终浓度的底物,使每孔100 μL,每孔3个重复。轻轻振板2 min,室温测定反应0 min、2 min和1小时时336/490 nm波长的荧光强度,探究底物消化的最佳酶浓度及反应时间。
结果分析:蛋白酶活性发挥需要时间。
对比两个时间点的荧光曲线可以找到某个蛋白浓度下在一定反应时间内所对应的荧光强度及荧光强度变化,即酶反应时间和快慢。为节约蛋白酶,理想的实验条件是在可控的短时间内具有较低的蛋白酶浓度及较为显著的荧光强度。
► 蛋白酶反应条件的优化
分别检测pH、金属离子、盐离子浓度及甘油浓度对蛋白酶活性的影响。反应中终浓度,蛋白酶为0.05 μM,底物25 μM,酶/底物=1:500 ,反应体积为100 μL。
pH对蛋白酶活性的影响:
|

配置pH 5.5~11.0的1 M缓冲液buffer 各50 ml,pH 值分别为 5.5,6.0,6.5,7.0,7.5,8.0,8.5,9.0,9.5,10,10.5,11。在黑色96孔板中加入各种pH条件下的基础酶切缓冲液和蛋白酶,离心混匀,设置酶标仪运行参数,再用排枪向96孔板中加入荧光底物。轻轻振板2 min,室温测定反应30 min 时336/490 nm波长的荧光强度,比较不同 pH 值条件下酶反应变化。
结论:Tris pH 8.5是该酶的最优 pH条件。
▲关键步骤:有些有颜色的缓冲液本身荧光背景较高,建议实验时改用无色透明缓冲buffer。
► 二价金属离子对蛋白酶活性的影响:
|

在黑色96孔板中分别加入终浓度为 0.1 /0.5/1.0 mmol/L MgCl2、CaCl2 、ZnCl2、MnCl2,再加入蛋白酶和基础酶切buffer,离心混匀,设置酶标仪运行参数,再用排枪向96孔板中加入荧光底物,轻轻振板2 min,室温测定反应30 min 时336/490 nm波长的荧光强度,比较不同浓度的不同二价离子对酶活的影响。
结论:不同金属离子表现出不同程度的蛋白酶抑制作用,浓度越高抑制越明显。
► 离子强度对蛋白酶活性的影响:
在黑色96孔板中分别加入终浓度为0 mM、50 mM、100 mM、200 mM 、400 mM和800 mM的NaCl,再加入蛋白酶,离心混匀,设置酶标仪运行参数,再用排枪向96孔板中加入荧光底物,轻轻振板2 min,室温测定反应30 min 时336/490 nm波长的荧光强度,比较离子强度对蛋白酶反应的影响。
|

结论:盐离子抑制蛋白酶活性,浓度越高抑制越明显。
► 甘油含量对蛋白酶活性的影响:
在黑色96孔板中分别加入甘油终浓度为 0%、5%、10%、15%、20%、25%、30%、35%、40%、45%,再加入蛋白酶,离心混匀,设置酶标仪运行参数,用排枪向96孔板中加荧光底物,轻轻振板2 min,室温测定反应30 min时336/490 nm波长的荧光强度。分析蛋白酶活性的变化。
 |
结论:低浓度甘油促进蛋白酶活性,在40%甘油时,酶活性最大。45%甘油表现出蛋白酶抑制作用。
总结:最佳酶切缓冲液是:50 mM Tris pH 8.5,40% glycerol,1 mM CHAPS。
► 蛋白酶米氏常数(Km)和最大反应速度(Vmax)的测定
96孔板中加入最佳酶切缓冲液和10 nM终浓度的酶,离心混匀,设置酶标仪运行参数,再用排枪向96孔板中加入不同终浓度的底物:0,1,2*2n μM(n=0~6),轻轻振板2 min,室温测定反应2 min时336/490 nm波长的荧光强度。计算反应初速度,根据米氏方程求得蛋白酶的Km 和 Vmax。
▲关键步骤:
米氏方程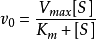 ,又称为Michaelis-Menten方程,是在假定存在一个稳态反应条件下推导出来的,其中
,又称为Michaelis-Menten方程,是在假定存在一个稳态反应条件下推导出来的,其中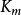 值称为米氏常数,
值称为米氏常数,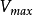 是酶被底物饱和时的反应速度,
是酶被底物饱和时的反应速度,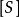 为底物浓度。双倒数作图法要注意选择数据尺度,不要使数据点太靠近原点。
为底物浓度。双倒数作图法要注意选择数据尺度,不要使数据点太靠近原点。
结果 | 米氏图 | 双倒数图 |
Vmax(FU/sec) | 32.25 | 20.03-116.9 |
Km(μM) | 35.51 | 13.45 |
R2 | 0.9870 | 0.9964 |
|

结论:同一组数据经米氏作图法和双倒数作图法得出的米氏常数(Km)和最大反应速度(Vmax)的差异较大。由于双倒数图的横轴截距为= - 1/ Km,纵轴截距为1/ Vmax,低浓度的实验点过度集中在零点,取其倒数后误差较大,对Km和Vmax的准确性影响较大。此情况解决办法,测定不同浓度蛋白酶时的Km。由于酶和底物的性质及反应环境不变,Km也理应不变。这里认为蛋白酶的Km =35.51 μM,Vmax =32.25 FU/sec(每秒钟的荧光单位变化)。
针对性建议(TROUBLESHOOTING)
不同蛋白酶特性不同,此文仅供参考。
3.5.2 ATP酶活检测
简介(INTRODUCTION)
ATPase/GTPase assay是用来测定具有ATP/GTP酶活性的相应酶的活性的方法。此类酶能催化ATP或GTP分解成ADP或GDP和游离磷酸盐。原理为:malachite green染料通过与游离磷酸相互作用产生激发光为620 nm的产物,且荧光强度与产物比例呈正比例。该方法迅速、高灵敏度。
材料(MATERIALS)
r 试剂(REAGENTS)
96孔板、带偏振光模块的酶标仪、锡箔纸、ddH2O、ATPase/GTPase assay kit
r 器械(EQUIPMENT)
水平离心机
► 试剂盒的配制
30 mL终止液的配制:0.035 g 孔雀绿,0.32 g钼酸铵,4.5 mL浓盐酸,25.5 mL ddH2O。
先加0.32 g钼酸铵,4.5 mL浓盐酸,25.5 mL ddH2O,室温搅拌至完全溶解后加入0.035 g 孔雀绿,室温搅拌一小时即可使用。该终止液可在一个月内稳定保存,最好是现配现用。确保使用的水不含游离的磷酸根。
Assay Buffer的配制:40 mM Tris, 80 mM NaCl, 8 mM MgAc2, 1 mM EDTA pH 7.5
实验步骤(PROCEDURE)
1. 基于ATPase/GTPase assay的原理,一定不能使用磷酸盐buffer(其他常见buffer兼容)且要确保各试剂及蛋白缓冲液中不含游离的磷酸盐,最好进行预测定。方法如下:加200 μL buffer到40ul样品溶液中,620 nm处的空白吸收值应小于0.3。如果吸光度数偏高,可能表示磷酸盐污染(如洗涤剂)。
2. 磷酸盐标准曲线的制定:用于比色检测的磷酸盐标准品为500 μL的50 μM的磷酸盐标准溶液。 将0,10,20,25,30和40 μL的50 μM磷酸盐标准溶液加入到96孔板中,相当于0(空白),500,1,000,1,250,1,500和2,000 pmol /孔(0,12.5,25,31.25,37.5和50 μM)的磷酸盐标准溶液。 向每个孔中加入超纯水使总体积达到40 μL。加200 μL buffer 测定620 nm处的吸收值。
3. 蛋白样品使用之前要离心以去除杂质,如果样品时组织或细胞则要用200 μL预冷的buffer稀释。最好进行不同蛋白浓度的预测定以确保620 nm的度数在标准曲线范围内,加入1~10 μL样品(预实验测定)至96孔板的重复孔中(三组重复)。使用Buffer将样品置于10 μL的最终体积。将10 μL的buffer作为阴性对照。
4. 加30 μL图1所示的Mix向每个实验样品,背景空白和阴性对照孔中,但标准孔中不加。
5. 在室温下孵育反应一段时间(例如30分钟),最佳时间可能需要通过实验确定。向每个孔中加入200 μL buffer,并在室温下再温育30分钟以终止酶反应并产生比色产物。为保证时间的同一,最好使用排枪。
6. 读取所有样品,标准品,空白和对照的600-660 nm处的吸光度[620 nm处的最大吸光度(A620)。通过从样品孔(A620)样品的A620中减去对照孔(A620)对照的A620,计算样品的吸光度值(A620)的变化。根据标准曲线得到实验组的磷酸盐浓度
Reaction Mixes |
|
Reagent | Samples,Background Blanks,and Negative Controls |
Assay Buffer | 20 μL |
4 mM ATP or GTP | 10 μL |

实例:
针对性建议(TROUBLESHOOTING)
1. 每次测定时,必须设置一个新的标准曲线。
2. 注意NTP 溶剂本身可能含有游离磷酸盐。
3.5.3 解旋酶活性检测
简介(INTRODUCTION)
解旋酶(Helicase)是生物分子代谢过程中关键的大分子物质之一,它广泛存在于从低等生物到高等生物( 病毒到人类) 多种生物体中。经过多年的研究,人们发现解酶具有多种重要生物功能,在细胞内它们参与DNA复制、修复、转录重组以及RNA接拼、核糖体组装、蛋白质翻译,端粒稳定,基因组稳定性等重要分子活动。

Heliacse的解链活性检测利用FRET(荧光共振能量转移)原理分析。其中一条链的5’端标记上荧光基团Cy3,另一条链的3’末端BHQ2作为猝灭基团。当解旋酶不存在时,DNA双螺旋结构稳定,由于荧光共振能量转移作用,此时体系荧光较弱。加入解旋酶后,由于酶对双螺旋DNA有解旋作用,使得双链DNA解离,导致末端标记的荧光基团与另一条链的淬灭基团远离,从而使体系的荧光增强。由于存在过量的第三条中和链,能够互补结合萃灭链,阻止其萃灭荧光链。
材料(MATERIALS)
r 试剂(REAGENTS)
Sample | Concentration |
Protein: helicase | 100 nM |
DNA substrate (Cy 3/B HQ2) | 50 nM |
“trap” DNA oligonucleotide | 500 nM |
MgCl2 | 1.0 mM |
ATP | 1.0 mM |
Buffer: 25 mM MES pH 6.5, 25% (v/v) glycerol, 0.1% Tween 20, 1 mM DTT | |
r 器械(EQUIPMENT)
酶标仪,96孔板(黑色不透明),EP管,PCR仪
实验步骤(PROCEDURE)
1. 等量的DNA单链放入Annealing buffer (10 mM MgCl2, 50 mM Tris pH 7.5)。DNA浓度越高越好,最好大于200 μM。90 °C 3 min,50 °C 3 min,20 °C 5 min即可退火,置于冰上。
2. 在含有1.0 mM ATP,1.0 mM MgCl2,50 nM 荧光标记dsDNA(5' Cy3/3' B HQ2)和500 nM与B HQ2链互补的捕获链的反应缓冲液(25% (v/v) glycerol, 0.1% Tween 20, 1 mM DTT, 25 mM MES pH 6.5)中,加入不同浓度的解旋酶,混匀,室温反应20 min,用荧光光谱仪进行扫描检测。
3. DNA 杂交后,550 nm波长激发Cy3基团产生的570 nm波长荧光被BHQ2基团吸收,因此对于双链DNA检测不到570 nm波长荧光光谱。当解链作用发生后,dsDNA分开,由于FRET作用的减少,550 nm波长激发Cy3基团,在570 nm波长处产生的荧光信号便逐渐增强。以同等浓度的5' Cy3单链在570 nm波长的荧光信号定义为100%双链完全解开强度(Fmax)。以双链在550 nm波长的荧光信号Fs定义为起始解旋值。解旋率(%)fraction unwound =( F-Fs) /( Fmax-Fs)。其中F值为加解旋酶检测到570 nm波长荧光信号强度。
 |
针对性建议(TROUBLESHOOTING)
荧光长时间曝光会发生荧光信号减弱现象,在实验过程中应避免过度曝光。
3.5.4 体外转录实验
简介(INTRODUCTION)
T7RNA聚合酶以dsDNA为模板转录产生ssRNA。其I型T7启动子序列为“5'GAAATTAATACGACT CACTATAG-3'”。转录从下划线的G开始(RNA产物第一个碱基为G),T7启动子下游第二位和第三位最好为G,使用互补链为模板。T7会在RNA产物3’末端加上一个或几个随机碱基。如果不理解看下图(下划线为T7启动子序列)
GAAATTAATACGACTCACTATAG XXXXXXXXXXXX
CTTTAATTATGCTGAGTGATATC YYYYYYYYYYYYY 产物为GXXXXXXX(加几个随机碱基)
为避免RNA产物3’端随机加碱基,可将模板链5’端的两个碱基2'羟基甲基化修饰,则RNA产物3’末端没有随机碱基。
二型T7启动子序列为:5'-TAATACGACTCACTATTA-3',复制从A开始,第二位和第三位最好为G。 TAATACGACTCACTATTA XXXXXXXXXXXX
ATTATGCTGAGTGATAAT YYYYYYYYYYYYY 产物为AXXXXXXX(加几个随机碱基)
材料(MATERIALS)
r 试剂
DEPC水、苯酚、氯仿、乙酸钠、无水乙醇、EDTA、NTPs、10×Transcription Buffer、尿素、丙烯酰胺、过硫酸铵、TEMED、0.5×TBE buffer、0.5×TB buffer、2×RNA native loading buffer、亚精胺、DNaseⅠ、NaCl
实验步骤(PROCEDURE)
1.线性化模板的准备
1) 用等体积的苯酚/氯仿1:1混合液提取DNA,必要时重复。
2)用等体积的氯仿提取两次以除去残留的苯酚。通过加入1/10体积的3 M乙酸钠pH 5.2和两倍体积的乙醇沉淀DNA。在-20 °C孵育至少30分钟。
3) 在微量离心机中以最快的速度将DNA沉淀15分钟。小心取出上清液。通过加入500 μL 70%乙醇冲洗沉淀并以最高速度离心15分钟。小心取出上清液。空气干燥颗粒,并以0.5~1 μg/ μL的浓度重悬于无核酸酶的水中。
4) 含有正确方向的T7 RNA聚合酶启动子的PCR产物可以被转录。在20 μL 体外转录反应中可以使用0.1~0.5 μg PCR片段。
▲备注:
♫ 底物DNA可以使用完全双链或带有双链T7启动子序列的单链寡核苷酸。但一般而言,单链模板产率相对较低。底物最好为线性(否则易滚环复制,模板双链DNA最好留下5’突出端)。
♫ T7对盐很敏感,DNA模板应该不含盐,虽然PCR混合物可以直接使用,但纯化的PCR产物可以获得更好的产量。确保DNA模板用酚/氯仿提取,用乙醇沉淀,溶于RNase free ddH2o中。
2.小体系摸索条件
体外转录RNA要对转录体系进行优化找到最高效率的转录体系。镁离子对于RNA的产率及产物的专一性影响很大,另外RNA体外转录所用模板和T7 RNA 聚合酶的量有时也对RNA 的产率有影响。镁离子浓度是必须要进行优化的,所以在进行大体积体外转录RNA前先进行 50 μL体系的小量转录优化镁离子浓度。
室温配置以下转录体系,要确保整个过程和体系中不含DNAse和RNAse。
| 终浓度 |
DEPC水 |
|
T7 RNA polymerase | 0.1 mg/mL |
RNA酶抑制剂(可选) |
|
DNA 模板 | 1 μg |
Mg2+ |
|
NTPs | 5 nM |
10× Transcription Buffer | 10 μL |
DTT(可选) | 10 mM |
| Total 50 μL |
1)在镁离子浓度优化时,一般设计 20 mM、25 mM、30 mM、35 mM、40 mM、45 mM、50 mM 共7 个梯度
2)37 °C孵育1~2 h(PCR仪),对于小于300 bp的孵育2~16小时。70 °C加热10 min或终浓度20 mM EDTA(8.0)可终止反应。
3)反应结束后14,000 rpm 离心去除焦磷酸镁沉淀或加EDTA溶解焦磷酸镁,随后进行Urea-PAGE或native PAGE凝胶电泳分析体外转录产量及RNA 条带和构象的专一性分析判断体外转录最优镁离子浓度。
▲ 备注:在T7RNA 聚合酶转录RNA 反应过程中会产生大量焦磷酸镁沉淀,可由此现象判断RNA 转录是否成功。
3. 凝胶电泳分析检测转录效果,尿素变性PAGE 或native PAGE 配置。
根据RNA的长度需选用不同浓度的聚丙烯酰胺配置的PAGE胶以达到最好的分辨效果有利于后续分析。RNA PAGE胶使用的聚丙烯酰胺为 40% 19:1 acrylamide:bisacrylamid贮存液。

一块变性PAGE胶的总体积为6 mL(选用尿素作为变性剂,浓度为7 M/L),
1)样品准备,转录完成的RNA 样品,离心或加EDTA 去除焦磷酸镁沉淀后稀释50~100倍(DEPC水),移液器取样10 μL于新的RNase free的EP管中,加入等体积2×RNA变性loading buffer,95 °C水浴锅或金属浴加热变形5分钟,立即置于冰上孵育5分钟,即可上样。对于native PAGE凝胶电泳的样品,操作与变性PAGE凝胶电泳样品一致,loading buffer为2×RNA native loading buffer。
2)相应电泳buffer尿素变性PAGE胶电泳buffer为0.5×TBE buffer,native PAGE电泳buffer为0.5×TB buffer。上样前恒压100~120 V预电泳30分钟,去除未聚合的丙烯酰胺及其他带电荷杂质。电泳过程中会产生热量,对于变形PAGE凝胶电泳来说,产热有利于RNA处于单链状态,但对于主要用于分析RNA构象的native PAGE来说,热量会导致RNA构象变化影响电泳结果分析,所以在跑native PAGE胶时,尽量低电压,在低温环境冰浴情况下进行。
3) 预电泳30分钟结束后即可上样。每泳道上样10 μL,Marker选用DNA Ultra-low ladder,虽然为DNA marker但可以大致估计RNA条带大小,特别是在10%PAGE胶时,其DNA marker条带与相对应长度RNA 条带基本一致,如果需要精确了解RNA 条带大小需用RNA marker。
4) 上样完成后继续恒压电泳,溴酚蓝指示剂条带的大小大概10 nt左右,根据目的RNA条带的大小待溴酚蓝指示剂跑到合适位置时停止电泳。小心剥离PAGE 胶置于培养皿中加入20 mL二级水。加入20 μL 1000 X gel red,摇床振荡孵育3分钟,二级水清洗去除未结合RNA 和非特异性结合胶的gel red,凝胶成像仪下观察拍照保存。RNA显示也可用0.1%的甲苯胺蓝,其操作过程与蛋白胶染色脱色类似,室温振荡孵育5分钟,水清洗几次,加过量水振荡孵育脱色或微波炉加热脱色。
▲ 备注:
♫ 转录需在无RNAse条件下进行。
♫ 反应体系在室温下配置,因为4 °C有亚精胺存在时DNA会发生沉淀。
4. RNA 大量转录
1)摸索好最优镁离子浓度后,即可扩大反应体系进行大量转录,以10 mL转录体积为例。向RNase free的50 mL 锥形管中依次加入下表试剂(pre-miR-1003镁离子最优浓度为30 mM):

混匀,37 °C水浴锅反应4小时。
2)反应结束后,加入200 U的RNase free的DNaseⅠ37 °C反应30分钟消化DNA 模板(也可省略)。
3)DNaseⅠ酶储存溶液:50 mM Tris-acetate pH 7.5,10 mM CaCl2,50%(v/v)glycerol。
4)Reaction Buffer(10×):100 mM Tris-HCl pH 7.5 at 25 °C,25 mM MgCl2,1 mM CaCl2。
5)反应体系置于70 °C水浴锅30 分钟使T7 RNA 聚合酶失活(如果3’端连接有HDV,则置于65 °C水浴锅30分钟,失活T7 RNA 聚合酶的同时提高HDV自切效率,5’端链接有hammer head ribosome RNA 则55 °C水浴锅20分钟)。
6) 待T7 RNA聚合酶失活后加入1/10 体积0.5 M EDTA溶解焦磷酸镁沉淀。加入1/10 体积5 M NaCl,高盐状态有利于后续的RNA乙醇沉淀。加入3倍上述总体积无水乙醇(RNase free预冷)-20 °C过夜沉淀RNA。
7) 在离心机4 °C 14,000 rpm离心30分钟,去上清,用70% RNase free乙醇缓慢清洗沉淀表面(去除沉淀表面吸附的高盐),敞口至乙醇挥发。加入1~2 mL DEPC 水溶解沉淀。加入等体积2×RNA变性loading buffer,90 °C以上水浴锅加热变形RNA 5分钟,随后冰浴10分钟等待上样。
8) 配置变性PAGE胶,纯化RNA 。
9)组装好胶板并用真空硅脂密封四周(注意密封完全)。不同浓度PAGE 胶配方如下:

10) 尿素溶解过程中吸热,加热促进其溶解。灌制PAGE胶时注意避免混入汽包。
11)待PAGE胶凝固后,0.5×TBE buffer,预电泳30 min,随后上样继续进行电泳(6 h)。溴酚蓝指示剂到合适位置,停止电泳,剥离胶板,用保鲜膜封闭 PAGE胶,黑暗中用手提紫外灯(波长245 nm)观察RNA 条带。
12)切下RNA条带,转入Elutrap电泳仪200 V 6小时或120 V过夜电洗脱(0.5×TBE buffer)。亦可以用“crush and soak”方法提取PAGE胶中RNA,RNA条带研碎加入0.3 M NaAC pH 5.3,2 mM EDTA浸泡,滤膜滤除PAGE胶颗粒收集上清液。
13)收集Elutrap洗脱的RNA不断加入2 M NaCl,浓缩管浓缩(高盐能去除结合在RNA上的小分子),加DEPC水浓缩去除高盐。在RNA低浓度情况下取出 95 °C 变性10 min,自然冷却或液氮冷却(变复性实验确保RNA 构象均一,冷却方法需摸索最适条件)。将RNA透析(浓缩)至后续实验buffer,浓缩至合适浓度-80 °C冰箱保存。
附录:所用试剂
以下所有buffer 都必须用DEPC 水配置灭菌或滤膜过滤
1.2×RNA 变性loading buffer:
95%(v/v)去离子甲酰胺,0.2%(2%?)(v/v)500 mM EDTA pH 8.0,0.02%(w/v)溴酚蓝。
2.2×RNA native loading buffer:80% 甘油,0.02%(w/v)溴酚蓝(可加可不加)。
3.1×RNA transcription Buffer:
1 mM DTT (或者多一点10 mM),40 mM Tris-HCl pH 7.9,6 mM MgCl2,2 mM spermidine(亚精胺)pH 7.9,0.5 mM each ATP、UTP、GTP、CTP(有的包括10 mM NaCl、0.05%吐温20)
4. 1×T7 RNA polymerase Storage buffer:
50% Glycerol,1 mM EDTA(有的0.1 mM),100 mM NaCl (或150 mM),0.1% Triton® X-100,50 mM Tris-HCl pH7.9,20 mM β-ME(巯基乙醇)pH 7.9,0.1 mg/mL BSA,(有的含有1或10 mM DTT)
5. 10×TBE buffer:
Tris 108 g,硼酸55 g,0.5 M EDTA,40 mL pH 8.0,蒸溜水定容至1000 mL。再将10×TBE稀释20倍成0.5TBE就可以在电泳时使用(即工作液浓度)
6. 0.5 M EDTA pH 8.0。
7. 18.61 g EDTA先加一定量ddH2O,用NaOH调pH值至8.0,再用ddH2O定容至100 mL,灭菌。
8. NTPs、 DTT、Tris、用DEPC水溶解。
9. EDTA、MgCl2、CaCl2用DEPC水配然后灭菌。
