5 结构生物学实验技术
5.5.1结晶筛选
5.5.2结晶条件优化
5.5.3晶体冻存
5.13.1 HKL2000 基本操作
5.13.2 CCP4基本操作
5.13.3 Phenix基本操作
5.13.4 Phenix.refine 常见问题和解答
5.13.5 Pymol 基本操作
5.13.6用Pymol创建动画
5.13.7用pymol和APBS画静电表面
5.13.8用Autodock做分子对接
5.1 圆二色谱
简介(INTRODUCTION)
圆二色光谱(简称CD)是应用最为广泛的测定蛋白质二级结构的方法,是研究稀溶液中蛋白质构象的一种快速、简单、较准确的方法。它可以在溶液状态下测定,较接近其生理状态。而且测定方法快速简便,对构象变化灵敏。样品对左、右圆偏振光(及正常的光)吸收程度不同,产生椭圆偏振光。在理解圆偏振光之前,首先要介绍自然光、平面偏振光、椭圆偏振光等概念。
自然光:光的一个固有特征是其振动平面与传播方向垂直,在自然光中,虽然每一束光的振动平面都与传播方向垂直,但不同束光之间的振动平面却无关联,可视为平均分配于垂直于传播方向的整个平面内,且平均来说,任一方向上具有相同的振幅,这种横振动对称于传播方向的光称为自然光既可将自然光看做圆柱形的光(下图)。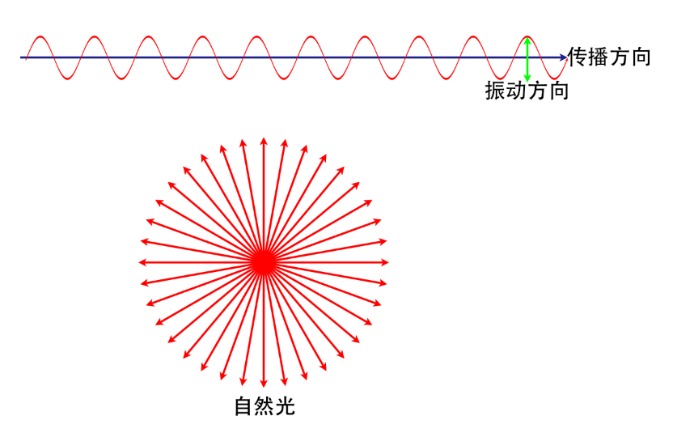
平面偏振光:如果在光的传播方向上光的振动平面在确定的平面内,这种光被称为平面偏振光或线偏振光。
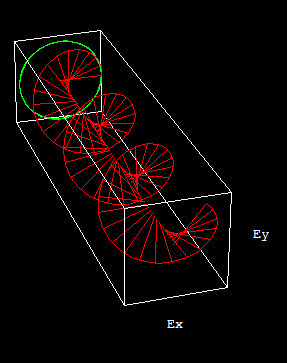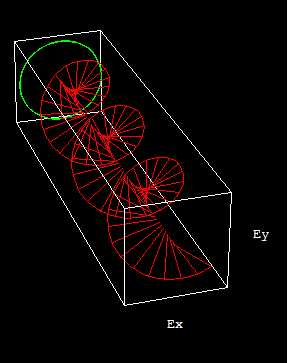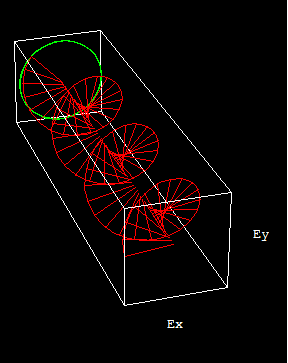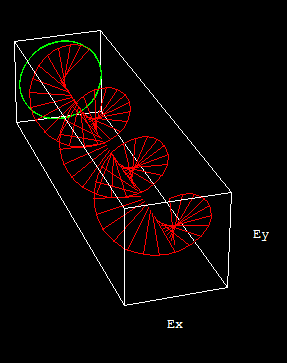
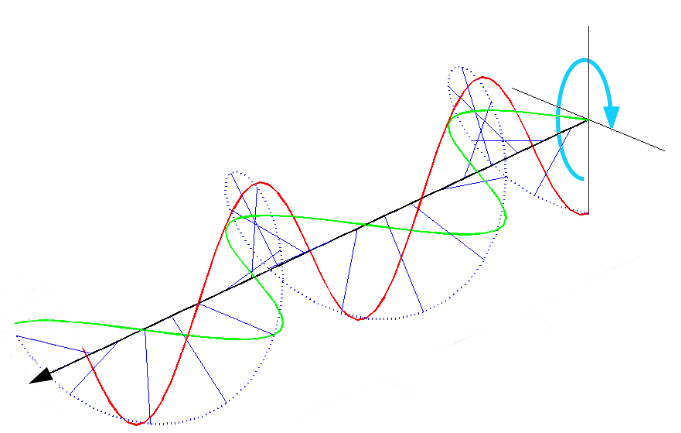
圆偏振光:如果光的振动平面随时间作有规则地改变,即振动平面轨迹在垂直于传播方向的平面上呈圆形或椭圆形,则称圆偏振光或椭圆偏振光可视为像弹簧一样的光,根据方向又可分左、右旋圆偏振光。因为光遵循矢量合成所以圆偏振光可视为传播方向相同,振动方向相互垂直且相位差恒定为1/2的两平面偏振光叠加后可合成光矢量有规则变化的圆偏振光。圆偏振光的电矢量大小保持不变,而方向随时间变化,即螺旋前进。
红绿为两束相互垂直,但相位差1/2的两条平面偏振光,蓝色虚线为合成的圆偏振光。
表格:不同光矢量合成效果
类型 | 频率 | 振幅 | 相位 | 合成效果 |
平面偏振光 | 相同 | 相同 | 差1/2 | 圆偏振光 |
左右圆偏振光 | 相同 | 相同 | 相同 | 平面偏振光 |
左右圆偏振光 | 相同 | 相同 | 不同 | 平面偏振光 |
左右圆偏振光 | 相同 | 不同 | 椭圆偏振光 |
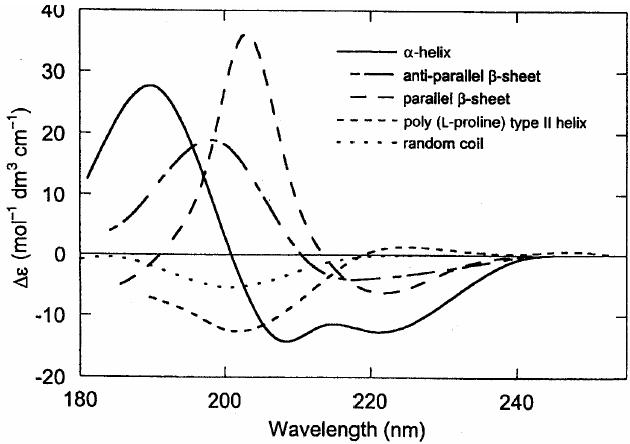
圆二色性基本原理:蛋白质或多肽中的光活性基团有肽键、芳香基团、二硫键。当平面偏振光通过这些基团时,其对左右圆偏振光的吸收不相同,造成左右圆偏振光的振幅不同,合成的圆偏振光变为椭圆偏振光。这就是蛋白质的圆二色性。在蛋白质分子中,肽链的不同部分可分别形成α-螺旋、β-折叠、β-转角等特定的立体结构。这些立体结构都是不对称的。蛋白质的肽键在紫外185~240纳米处有光吸收,因此它在这一波长范围内有圆二色性。几种不同的蛋白质立体结构所表现的椭圆值波长的变化曲线──圆二色谱是不同的。如下图所示,α-螺旋的谱是双负峰形的,β-折叠是单负峰形的,无规卷曲在波长很短的地方出单峰。蛋白质的圆二色谱是它们所含各种立体结构组分的圆二色谱的代数加和曲线。因此用这一波长范围的圆二色谱可研究蛋白质中各种立体结构的含量。
利用圆二色谱验证蛋白质稳定性
通常实验中需要验证蛋白质与小分子结合或者蛋白质本身的稳定性,利用圆二色谱测定蛋白质(与小分子结合)的变温曲线,我们可以通过计算其Tm值确定其稳定性。
材料(MATERIALS)
试剂(REAGENTS)
石英杯清洗剂(仪器中心提供)
缓冲液(BUFFER)
磷酸盐缓冲液,150 mM NaCl
器械(EQUIPMENT)
Chirascan Spectrometer (Applied Photophysics,Leatherhead,UK)、1 mm厚度的石英比色皿
实验步骤(PROCEDURE)
1. 准备蛋白样品,浓度为0.3~0.5 mg/mL,蛋白所在缓冲液为磷酸盐缓冲液;
2. 圆二色谱仪开机后需要充氮气约30 min,再打开疝气灯。设置扫描波长范围:180 nm~260 nm,重复2次。
3. 先检测仪器稳定性,直接扫描空气,得到一个范围在0.1附近的直线为正常,在检测蛋白圆二色谱曲线前分别检测对应的缓冲液曲线(应该是一条直线);
4. 检测样品二级结构折叠情况:吸取200 μL蛋白样品至规格为1 mm厚度的比色皿中,透光观察避免气泡,温度为20 °C,扫描后可得到曲线;
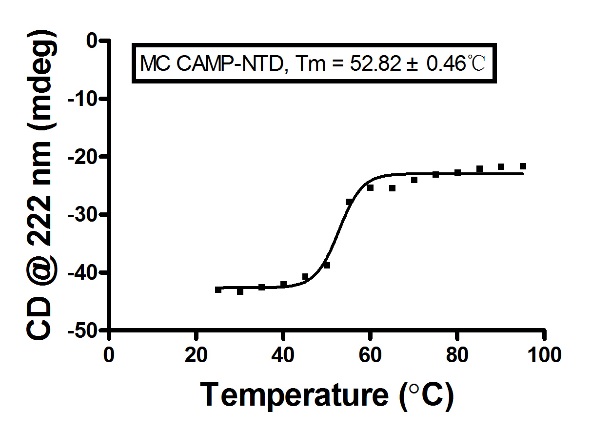
5. 检测蛋白质热变性:需要开启冷循环控制器。吸取200 μL蛋白样品至规格为1 mm厚度的比色皿中,透光观察避免气泡,将温度探测针插入样品中,温度范围是20 °C~95 °C,每隔5 °C检测一次,每次检测重复2次。根据蛋白初始二级结构光谱曲线取220 nm或者222 nm处的数据用GraphPad Prism 5.0做图,拟合并计算Tm。
附:数据处理与结果
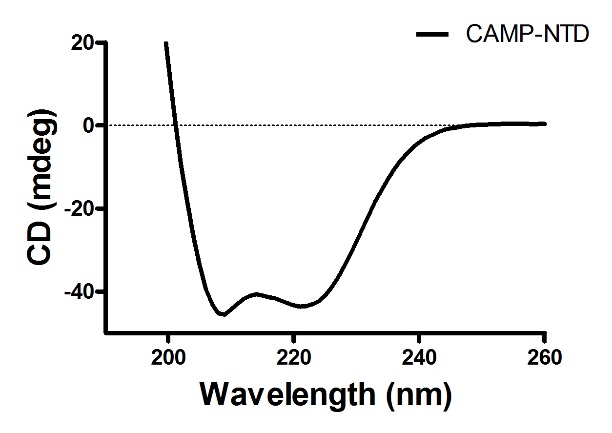
验证Mobiluncus curtisii CAMP-NTD的二级结构及其热稳定性。
1. NTD的圆二色谱:经如上实验步骤获得实验数据,导入Prism中作曲线图,得如下结果,为规则的α-螺旋:
2. NTD的热变性曲线:选取EXCEL数据表中222 nm处的数值(为不同温度在222 nm扫描得到的数值)导入Prism中作曲线图,得到如下结果:
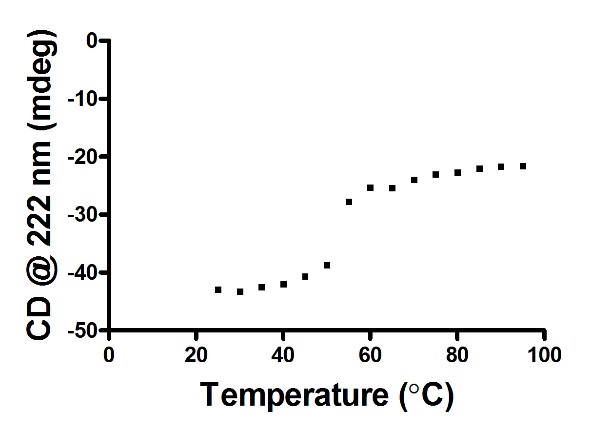
3. Tm值的计算:
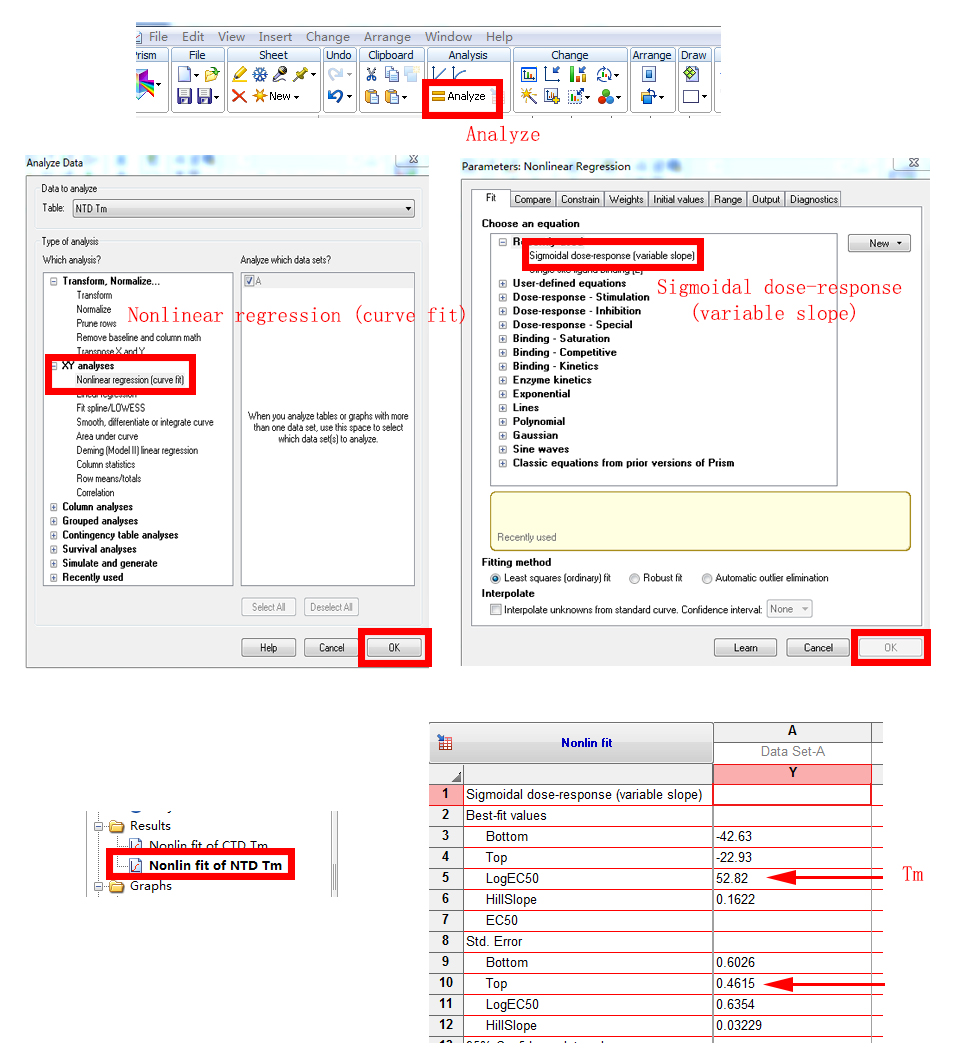
4. Fit后的结果,如下图:
针对性建议(TROUBLESHOOTING)
样品要求:
1. 样品必须保持一定的纯度不含光吸收的杂质,溶剂必须在测定波长没有吸收干扰;样品能完全溶解在溶剂中, 形成均一透明的溶液;
2. 氮气流量的控制,实验中途要时刻关注氮气是否充足,如不充足需及时更换氮气瓶;
3. 缓冲液、溶剂要求与池子选择:缓冲液和溶剂在配制溶液前要做单独的检查,看是否在测定波长范围内有吸收干扰,看是否形成沉淀和胶状;在蛋白质测量中,经常选择透明性极好的磷酸盐作为缓冲体系;
4. 样品浓度一般在0.05~0.5 mg/mL,浓度太高噪音太大会影响结果;
5. 样品不同,测定的圆二色光谱范围不同,对池子大小(光径)的选择和浓度的要求也不一样;蛋白质CD光谱测量一般在相对较稀的溶液中进行;
6. 保持石英杯干净透亮,通常完成蛋白热变性检测后石英杯内部会有蛋白质粘物,需要用清洗剂浸泡30分钟,再用清水冲洗干净;
7. 用去垢剂洗完石英杯后,需要用70% 酒精进一步清洗。
5.2 小角散射
简介(INTRODUCTION)
X光散射技术是常用的非破坏性分析技术,可用于揭示物质的结构、化学组成以及物理性质。这些技术是以观测X射线穿过样品后的散射强度为基础,根据散射角度、极化度和入射X光波长对实验结果进行分析。
散射包括弹性散射和非弹性散射,弹性散射包括小角X射线散射(SAXS)、广角X射线散射(WAXS);非弹性散射包括康普顿散射、共振非弹性X射线散射及X射线拉曼散射。SAXS主要测量散射角2θ接近0° 时的经过样品后的X射线散射强度,而WAXS是测量散射角2θ大于5°。
在原理上散射振幅等于电子密度的傅立叶变换乘以一个角度相关的因子。假设样品有很多一样的颗粒组成,每个颗粒里面的电子密度以ρ(r)表示,最大的维度为Dmax,那么总的散射强度可以写成球坐标形式:,其中γ(r)是密度的自相关函数的球形平均值(同一个长度,不同方向的平均)。I(s)的极限即为Guinier公式,也就是ln[I(s)] vs s2,这个极限公式在s<的范围内适用,Rg为回旋半径。
SAXS的优点:对样品的要求很低,溶液样品即可,对分子量和浓度没有要求。由于SAXS在溶液中进行,因此更好地反应生物大分子的真实状态,对原位研究动态过程提供了可能性。
SAXS的缺点:得到的信息量很少,要得到三维结构的信息很困难,只能得到一些比较粗略且低分辨率的信息,如生物大分子的大小、形状、某些关键的片段、各个组分之间的空间关系等。对于SAXS来说,分子越大实验越容易,这一点和晶体学正好相反。
SAXS数据分析先用RAW软件:
(可以从http://www.macchess.cornell.edu/MacCHESS/RAW_install.html免费下载软件并根据说明进行安装)来进行data reduction之后,一般用ATSAS软件包,里面包括很多小软件,根据不同的需要选择合适的软件。求解大体形状时可以用gnom、damin、gasbor等软件;如果是重建柔性区域可以用credo、corel、crysol等软件;如果是复合物结构重建则需要用massha、sasref、crysol等软件。
每一种软件都可以在https://www.embl-hamburg.de/biosaxs/software.html上活得对应的manual。
材料(MATERIALS)
试剂(REAGENTS)
60 μL高纯度蛋白样品(浓度在1~10 mg/mL范围内、根据分子量、分子量越小浓度需要高一些、分子量越大浓度需要低一些),如果是胞内蛋白可以加2~10 mM DTT作为radiation damage保护剂。
10 mL严格对应的buffer
器械(EQUIPMENT)
同步辐射加速器19U2线站
实验步骤(PROCEDURE)
Data reduction
1. 将蛋白在1~10 mg/mL之间用对应的buffer稀释三种梯度,并放在线站内的样品托盘上。
2. 在线站的软件上设置样品的名称,收集顺序以及保存路径。
关键步骤:这里需要注意的是,该软件是在linux环境下工作的,因此在样品名称中不得出现空格和/等符号,如果一定要用分割则用“_”下划线符号。还有,如果是在新的文件夹中开始收集,在Next tube No.上填写“1”,如果是在原来的路径中继续收集的话千万不要更改,不然同一个序号的数据后者会覆盖前者,这就意味着之前收集的数据需要重新收。
3. 打开光源,并点击“run”,开始收集数据,在线站的ALBRA软件上检查detector收集的散射光图片是否正常,如果是圆形的光斑那就是正常的,如果边缘上有刺头那说明光路有问题,需要找线站的工作人员重新调整光路。
4. 收集好的数据传到线站中的另一台Windows电脑上,此电脑上已经安装好了RAW、ATSAS等分析软件。每一次收数据之前,线站的工作人员会帮我们调整好光路和设置,并保存一个cfg文件,打开RAW,先将路径调整至cfg文件所在的位置,并双击该文件,这样才可以导入当天的参数。
5. 然后将文件过滤成tif文件,每一个样品我们一般收集20张数据,选择某一个样品的20个数据,点击“plot”,此时旁边的窗口会显示20个曲线并且正常情况下会几乎重叠在一起,如果没有,将不重叠的那些数据删除,剩余的数据选择后点击“average”。平均出来的数据我们需要自行保存。
6. 一般每一个样品前后都会测对应的buffer,因此可以选择前面或者后面紧挨着的数据作为该样品的背景并扣除。将样品和buffer都进行平均后,在buffer数据旁边点亮“★”,并选择样品,点击“substract”。此时得到的数据是将背景扣除后的样品纯粹的信息。同样,substract后的数据也需要保存,一般文章中提供的原始数据都是从substract开始展示,并且后面的一系列分析也都是基于这个原始数据来进行的。因此这一步要做好,不然后面的分析都不可信。
7. substract后的数据可以用记事本打开,里面的数据可以用prism去重新绘图。
Data analysis
1. 打开ATSAS软件中的“SAS Data Analysis”,将通过data reduction得到的substract数据拖到界面中。
2. 一般比较三种浓度梯度的信号强度,保证s小的地方信号没有过度的条件下s大的地方信号尽可能少的波动。如果数据从头到尾都是信号很强的状态,那么这种数据偏artifitial。如果三种浓度梯度信号没有一个能满足这个条件,可以将高浓度的尾部和低浓度的头部进行merge,方法是通过软件中的“processing”中的“scale”将高低浓度的数据进行拟合,使两个数据在某一点有重叠,再点击“merge”,这时软件会自动生成一个新的数据,而这数据可以用来后面的进一步分析。
3. 初步分析,一般需要看样品的回旋半径是否是浓度依赖的?Dmax是不是浓度依赖的?
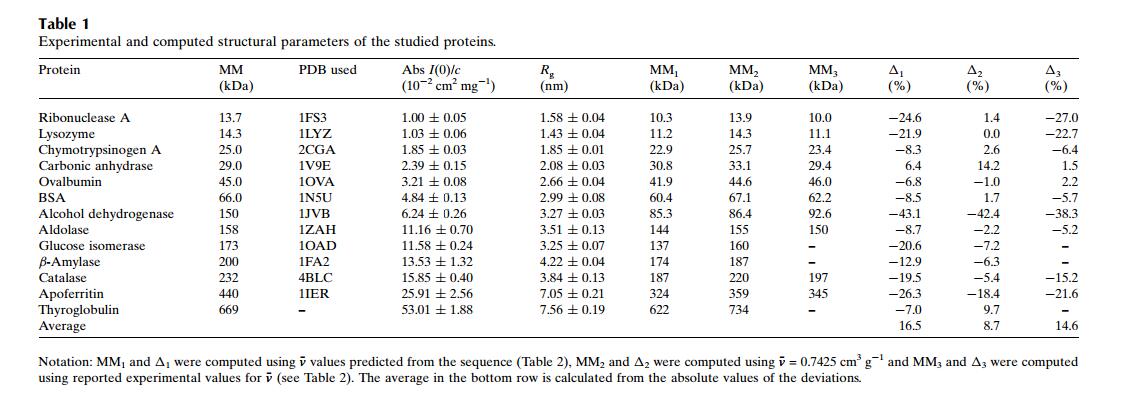
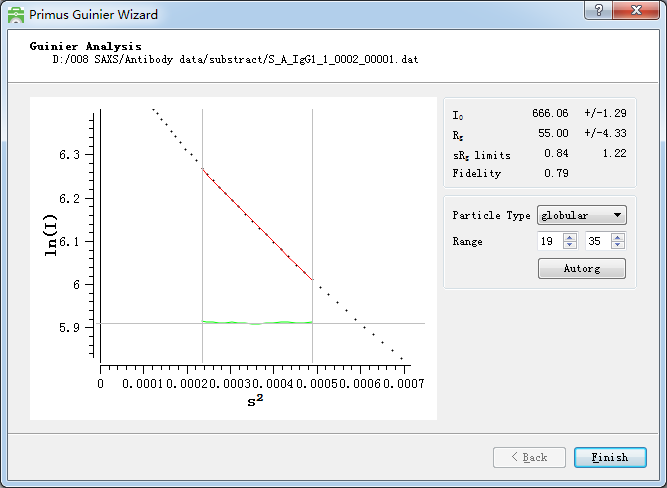

1. 因为回旋半径就跟蛋白的等电点一样是样品的固有属性,原则上它不可能随着样品的浓度的变化而变化。Dmax是样品的最大直径,如果溶液中的样品是一个均匀的介质,那么这个也不会是浓度依赖的。如果Dmax随着浓度高而变大,那么极有可能是样品有很强的聚集能力,这种情况下我们只能用低浓度的数据。这里放一张回旋半径和对样的分子量的表格,回旋半径和分子量是呈正比的,分子量越大回旋半径也越大。回旋半径可以通过软件中的“radius of gyration”即回旋半径,来进行计算,这里用到的是guinier公式,因此需要满足,并且需要让实际的样品曲线尽量和拟合后的线性曲线重叠。同时也要保证绿色的曲线的上下分布是均匀的,避免多数在上面或者下面。
2. 下面是Dmax的计算,点击“distance distribution”,会出现如下图。我们需要关注的是p(r) vs r曲线的尾部需要平缓地往下走,如果是很陡的那种需要人为地在range中改变包括的点的数目来变化。上面的quality是表征这套数据的质量的标准,当然,这个数值越高越好,但我们更多还是要看圆圈中的图形。点的数目不需要太多,如果好的数据可以留很多点,但如果s很大的地方噪音很高的话,可以只留400~500个点,即到s在0.2左右的数据也是可以的。点击“finish”后会提醒保存数据,保存后可以用记事本打开,将里面的数据用prism重新画图。
Modeling
1. 做完初级分析后,后面完全是根据实验目的来选择特定的软件,在SAS Data Analysis中已经整合了dammif、crysol、oligomer、Bodies等常用的软件。其他的软件全部都可以在ATSAS软件包中能找到,不同的软件在Embl-hamburg网站上都能找到manual,因此这里用dammif来介绍如何建模。
2. 点击“dammif”,选择“manual”,在计算Rg中选择合适的点,在Dmax上参考第11步,也可以在前面把数据记下来,直接在这里输入,点击“next”,直到最后界面上。如果直到该样品的对称性可以选择,如果不知道默认是P1对称性。在anisometry上可以选择是球形还是长棍形,以及在angular scale上可以选择是nanometer还是angstrom级别的,默认都是unknown。在mode上可以选择fast或者slow,区别是计算时间上fast更快,并且模型中球的数量更少,但轮廓更明显。如果是slow模式的话,计算时间很长,模型中球的数量更多,轮廓不明显。在repetition上可以选择计算的轮数,可以自定义计算几轮,每一轮的计算产生一个模型,最后通过damaver和dammin进行refine后可以通过RMS比较来选择最优的值。

针对性建议(TROUBLESHOOTING)
1. 首先要明确实验目的,小角散射不像晶体衍射能解释到原子层次上,不同的目的需要用到的软件也是不一样的,如果只是为了凑数据,那么我还是建议大家把更多的心思放在长晶体上,不要觉得小角散射是个投机取巧的捷径。大部分情况下,小分子(小于80 kD)且单体蛋白不适合做小角散射,前面也提到了蛋白越大越好,当然也不能是aggregate。如果是有规则的多聚体,那么可以用WAXS。
2. 其次,小角散射一般是辅助型实验,比较适合与晶体结构或者NMR联用,如果只是单纯SAXS数据因为其分辨率很低的因素,无法得到具体的结果,但有一点需要强调的是,它是检测蛋白在溶液中的构象的很方便的手段。因为同是溶液样品,NMR只能用在很小的样品,而SAXS在这一点没有什么限制。
3. 如果只要确定蛋白的形状,可以只去前面的数据,不需要取那么多数据。
5.3 负染
简介(INTRODUCTION)
负染就是用重金属盐(如磷钨酸、醋酸双氧铀)对铺展在载网上的样品进行染色;吸去染料,样品干燥后,样品凹陷处铺了一薄层重金属盐,而凸的出地方则没有染料沉积,从而出现负染效果。可以显示生物大分子、细菌、病毒、分离的细胞器以及蛋白质晶体等样品的形状、结构、大小以及表面结构的特征。
材料(MATERIALS)
试剂(REAGENTS)
2%或者3%醋酸铀
器械(EQUIPMENT)Gaten plasma systern、铜网、精细镊子、格式化的U盘、Tecnai G2120kv电镜

实验步骤(PROCEDURE)
一、 Tecnai G2120kv电镜基本操作步骤
1. 准备步骤
向冷阱中加液氮,如右图所示:电镜在在使用之前需要提前冷却,冷阱冷却镜桶大概需要1小时以上。
(1)进入电镜控制系统(操作者需要进入自己的User账户),检查电脑屏幕右下角托盘中如右图图标是否为绿色(![]() ),按顺序启动,按顺序启动TUI(Tecnai User Interface)和TIA(TEM Image and Analysis)系统。
),按顺序启动,按顺序启动TUI(Tecnai User Interface)和TIA(TEM Image and Analysis)系统。
(2)升高压,点High Tesion,将高压升至120 kv。
(3)开灯丝,点Filament(注:当长时间离开时,需关闭灯丝)。
2. 准备样品
常规常温样品准备
(1) 制备带有样品的铜网。
用Gatan plasma system等离子清洗机处理铜网,氢气氧气处理10秒。
将自锁镊子夹住铜网,正面朝上,加5 μL样品,静置1 min吸附。用滤纸边缘吸去多余样品,加5 μL醋酸铀,静置1 min染色。用滤纸边缘吸去多余染液,灯下烤干15 min以上。
注:样品多且对温度不敏感时,可在封口膜上多个样品同时操作。样品可保存一周。
(2) 按下图将样品铜网固定在样品杆上。

(3) 取出下图红色箭头处的工具。

(4) 使用该工具将样品杆末端的弹簧夹掀起,用镊子把样品铜网放入样品。
注:铜网插入电镜前,必须完全干燥。
3. 插入样品杆
常温常规样品杆
(1)设置抽真空时间:Vacuum>setting>pumping time>120 s
(2)检查镜筒阀是否关闭,黄色(Col.Valves Closed)表示关闭。Setup>Col.Valves Closed

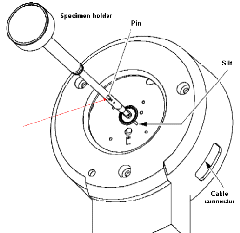
(3)将样品末端的细小针尖(下图中红色箭头所示位置)对准样品台上的细缝(五点钟位置),插入样品杆。预抽循环将会自动开始,请等待直至样品台上红色指示灯熄灭。下图箭头所示红灯熄灭后,将样品杆逆时针旋转至少十二点钟位置,然后小心缓缓将样品放入。
(4)检查设置页中镜筒真空读数,即Column值是否为20以下,若在20以下,即可打开镜筒阀,点击“Col.Valves Closed”按钮,此时V4和V7阀会被打开,即可开始观察样品。
4. 电镜基础调节
(1)Eucentric Focus
做alignment前,必须按下Eucentric Focus,如下图所示:用Intensity改变光斑大小的时候,最好顺时照明(即顺时扩大光斑)


(2)调节Z-high,使样品位于Eucentric height
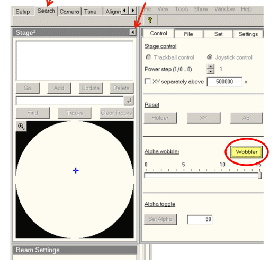
寻找样品中一特定物体作为参照物,激活Alpha Wobbler,样品台正负15°摆动,调节Z轴按钮使荧光屏上的目标物近似不动,如下图所示:

(3)Gun tilt
激活Direct alignment中的gun tilt 功能后,用Multifunction X/Y旋钮将荧光屏上的电流值(screen current)调到最大,肉眼观察是调至光斑最亮时。
(4)C2聚光镜光栏对中及象散矫正
A. 聚光镜光栏居中,插入聚光镜光栏(一般生物样品用3号光栏),
Intensity逆时针聚拢电子束,Beam Shift移光到中心,顺时散开电子束,若光斑与荧光屏不是同心相切,则调节C2光栏上的X/Y(如下图所示)旋钮使光斑同心相切。

重复以上步骤,保证光束于最小和最大状态下都位于荧光屏中心。
B. C2聚光镜象散矫正
若光斑呈椭圆形,则说明C2聚光镜有象散,需要矫正选择Stigmator功能(如下图),点Condenser。
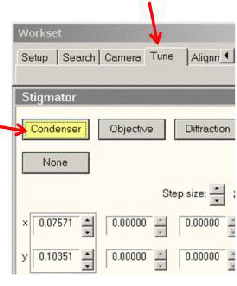
如下图,使用面板中的多功能按钮,调节X和Y方向上的象散校正线圈的强度,使光束变圆并且能够同心散开。

C. 点击调节页中的None按钮,结束象散调节。
(5)Direct alignment
如下图所示,转到用户界面中的调节页,选择直接调节项目。
A. Beam tilt pivot point
依次选择下图中红框内的每一项,使用控制面板上的多功能调节旋钮,使荧光屏中的两束光重合,且颤动最小。
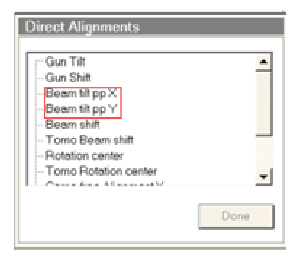
B. Beam shift
选择Beam shift项目,使用多功能调节旋钮,将光束移动至荧光屏中央。
(6)物镜光栏居中及物镜象散矫正
A. 物镜光栏居中
在diffraction模式下插入物镜光栏(一般生物样品用三号光栏),调节物镜光栏上的X/Y旋钮将物镜光栏居中。

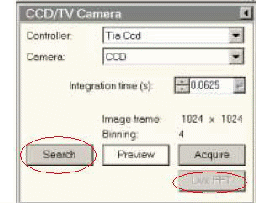
B. 物镜象散矫正
在有碳膜区域,稍欠焦状态下,CCD观察,如上图,激活Live FFT,Stigma→objective→MF X/Y将傅立叶环调成圆形。
(7)Gain reference(选做,做ET必须要做的一步)
A. 去除暗电流:
(a)找一个空白区域,光散开。
(b)关掉column。
(c)CCD/TV Camera→Bias gain reference→All Bias。
B. Gain reference
(a)开column。
(b)在空白区域,光散开至整个荧屏。
(c)点击All gain。
C. 检测gain reference的效果
(a)Acquire一张图。
(b)点击TIA软件右边的Auto-Correlation(做的好的状态是只有中间有一个亮点)。
注:做冷冻电镜时,上述的电镜基础调节步骤需要在Exposure模式下做
5. 样品观察
low dose
(1)选定Search、Focus和Exposure所需要的放大倍数及Spot size及所需要的光斑大小。
(2)Search模式与Exposure模式位置对准,找一个明显的样品参照物,在Exposure模式下用样品扭杆将参照物移到视眼中央,在切换到Search模式下,点击low dose→option→search shift,调节MF X/Y,将参照物移到视眼中央。
(3)在Search模式下寻找样品,Focus模式下调节焦距,Exposure模式下拍照。
5.4 冷冻电镜样品制备
简介(INTRODUCTION)
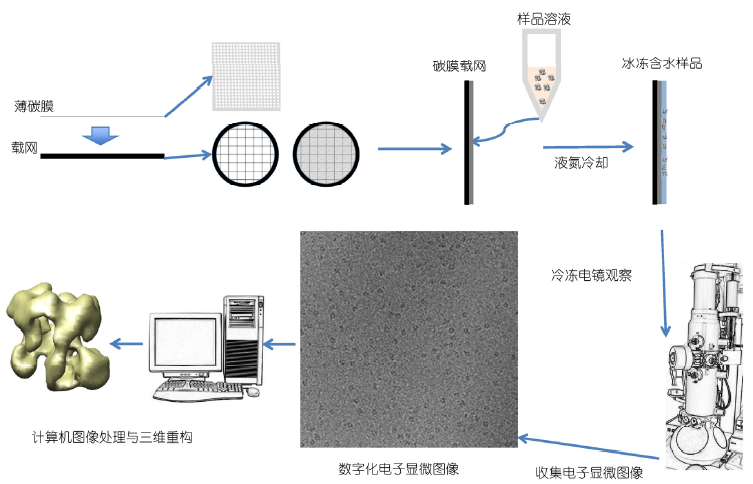
在低温下使用透射电子显微镜观察样品的显微技术,就叫做冷冻电子显微镜技术,简称冷冻电镜(cryo-electron microscopy,cryo-EM)。
冷冻电子显微学解析生物大分子及细胞结构的核心是透射电子显微镜成像,其基本过程包括样品制备、透射电子显微镜成像、图像处理及结构解析等几个基本步骤(图1)。在透射电子显微镜成像中,电子枪产生的电子在高压电场中被加速至亚光速并在高真空的显微镜内部运动。根据高速运动的电子在磁场中发生偏转的原理,透射电子显微镜中的一系列电磁透镜对电子进行汇聚,并对穿透样品过程中与样品发生相互作用的电子进行聚焦成像以及放大,最后在记录介质上形成样品放大几千倍至几十万倍的图像,利用计算机对这些放大的图像进行处理分析即可获得样品的精细结构。
样品需求量少:一个冷冻样品只需要3~5 μL 0.1~1 μmol 的蛋白质溶液;更接近生理状态:冷冻电子显微学通过将样品快速冷却至玻璃态冰达到固定生物含水样品的目的,其观察的结构信息基本上反映样品冷却前的瞬时状态;适用研究对象广泛:从细胞、细胞器到分子量在200 kD以上(最近的一些工作报道了分子量在200 kD以下的蛋白质分子的冷冻电镜结构)的大分子复合体。
材料(MATERIALS)
器械(EQUIPMENT)
Vitrobot
实验步骤(PROCEDURE)
Vitrobot制样基本操作
1. 加水:用针筒从下面软管注入纯水,大约20~30 mL。
2. 装滤纸:打开机器,装上滤纸,点击Resetblotpaper。Blot污点。
3. 设置参数
(1)Console下设置:温度(一般设置为22 °C),湿度(设为100%)
(2)Options下设置:
A. Miscellaneous:
选上“Use Footpedal”、“ Humidifier Off during Process”、“ Skip Grid Transfer”
B. Process paprameter下设置以下参数:
Blot time(S):滤纸吸附铜网液体时间
Blot force:滤纸夹铜网的力度
Wait time(S):吸附前的等待时间
Blot total:滤纸吸附次数
Drain time(S):Blot后的等待时间
Skip application:跳过加样
4. Ethane Container准备工作
从中间的孔加液氮,充满整个Ethane Container,让其冷却。等中间孔中液氮挥发完后再向孔中加少许液氮再次冷却,同时将孔外面液氮加满。等孔中的液氮完全挥发后开始通乙烷,乙烷加八分满。待乙烷固液共存状态时移走导热杆。
注:等孔中液氮完全挥发后开始通乙烷。液氮干净新鲜,液氮罐干燥。
5. 制样
(1)装镊子
镊子夹好铜网后(铜网需事先做glow discharge),装在vitrobot上,让铜网正面朝右,镊子有字一面朝向操作者。踩一下脚踏板,将镊子升上去。
注:铜网要夹紧
(2)将Ethane Container放在操作台上,踩一下脚踏板,将Ethane Container升上去。
(3)打开Humidity,将湿度升到100%后关掉。
(4)踩一下脚踏板,镊子掉下一点,加样品。
(5)踩一下脚踏板,滤纸吸附多余样品→镊子快速掉进乙烷中→Ethane Container降下来
(6)补充液氮,取下镊子。注意不能碰撞铜网,也不能将铜网离开乙烷。
(7)将Ethane Container转移至桌面,松开镊子上的固定圈,将铜网迅速转移到液氮中然后转移至样品盒内。
6. 收尾工作
取出滤纸,镊子用电吹风吹干,将Container中的液氮和乙烷倒掉后放入通风橱风干,退出程序关机,关闭Vitrobot的电源。抽出剩余的纯水。将所有物品归位,登记使用记录。
5.5.1 结晶筛选
简介(INSTRUCTION)
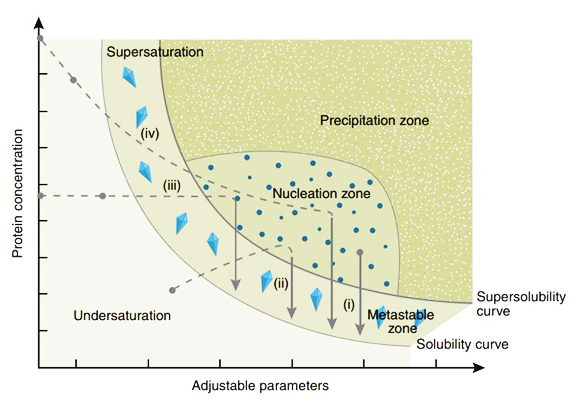
蛋白结晶是一个有序化过程:即蛋白质由在溶液中随机状态转变成有规则排列的状态。当蛋白质溶液达到过饱和状态时,能够形成一定大小的晶核,溶液中分子失去自由运动的能量,不断结合到形成的晶核上而长成适合 X 射线衍射的晶体。结晶过程分为两步:首先形成晶核,而后形成晶体。其中形成晶核是一个关键的步骤。以蛋白结晶的相变过程简要介绍结晶的一般方法以及涉及到的关键因素。有4种主要的蛋白结晶方法:(Ⅰ)批量结晶法(Microbatch);(Ⅱ)气相扩散法(Vapor Diffusion);(Ⅲ)透析法(Dialysis);(Ⅳ)自由界面扩散法(Free Interface Diffusion)。尤以气相扩散法最为常用,又细分为悬滴法(Hanging Drop)和坐滴法(图Sitting Drop)。蛋白浓度、沉淀剂浓度、添加剂浓度、pH、温度等都是影响蛋白结晶的关键因素。这里以气相扩散法来说明蛋白结晶的相变过程(图Phase Diagram),假定浓缩的蛋白溶液与母液以一定比例混合后形成的液滴刚开始是澄清的,也就是说蛋白质分子尚处于非饱和状态,由于混合的液滴与下槽的母液都处于封闭环境中,并且母液的浓度要高于液滴的浓度,因此随着时间的延长,借助水蒸气的扩散,液滴的水分会逐渐减少,意味着其中的蛋白质浓度与沉淀剂浓度都会逐渐升高,直到条件变化至Nucleation zone中,晶核形成,随后溶液中的蛋白分子不断自发地结合到形成的晶核上从而长成适合X射线衍射的晶体,即相变至Metastable zone Nucleation zone至Metastable zone 的过程中可以看到液滴中的蛋白浓度是直线下降的。

材料(MATERIALS)
试剂(REAGENTS)
目的蛋白、结晶板、凡士林、硅化盖玻片等耗材
器械(EQUIPMENT)
恒温培养箱、体视显微镜
实验步骤(PROCEDURE)
1. 评估蛋白样品:
浓度范围:5~25 mg/mL;MBP 融合蛋白尽量浓缩至 30 mg/mL,甚至50 mg/mL以上。一般水溶性好的蛋白,初始浓度要求高一些。分子量小的蛋白,浓度要求高一些。分子量大的的蛋白,浓度要求低一些。在纯化到目的蛋白之后,点晶体之前,需要对目的蛋白做尽量多的状态评估,以提高实验效率。通常蛋白状态评估常见问题。
(1)蛋白纯度。纯度是结晶性能最重要的前提条件。纯蛋白意味着翻译后修饰不存在异质性,也意味着杂质占总蛋白质的含量较低,如1%。可通过运行一块过载的凝胶电泳来检测蛋白纯度。如果要结晶蛋白-蛋白复合物,在建立结晶前需要进一步纯化,使形成复合物的蛋白从未形成复合物的蛋白中分离。
(2)蛋白折叠。可检测蛋白活性,也可检测蛋白的圆二色谱(CD)来反应蛋白是否正确折叠。
(3)新鲜制备蛋白。蛋白会随着时间降解使混合物变得不均匀。最好在蛋白纯化完成的当天进行结晶实验。
(4)单一的蛋白聚合状态。如仅存在单体或二聚体的任意一种。可将分子筛纯化方法作为蛋白纯化的最后步骤。可使用动态光散射器确定蛋白的聚合状态。
(5)蛋白浓度。
(6)蛋白在室温下是否稳定。是否需要添加一些东西(如盐)。蛋白降解是否迅速。
(7)类似的蛋白是否结晶过?检查PDB并查看头部记录以获得结晶化细节。
2. 选择适当的晶体筛选试剂盒。
市售多种蛋白结晶筛选试剂盒,大多是基于随机法或者不完全因素法(Sparse matrix screen)设计的。最早由McPherson报道,收集了最常见的蛋白结晶条件,把这些因素作随即组合而成。最经典的就是Hampton Research 的Crystal Screen Kit。后来新开发的试剂盒,参考了更多蛋白样本。还有一些有一定针对性的试剂盒,比如针对蛋白复合物的试剂盒,Protein Complex Screen, 由NIAID Peter Sun组设计。Hampton Research公司的Natrix Screen针对核酸蛋白复合物的结晶。TJ-Lab有常见的10多种结晶试剂盒,可以根据需要和样品特性选择。
3. 蛋白样品需要选择适当的缓冲液。
需要对蛋白缓冲液的喜好有一定的了解。比如,缓冲液pH远离蛋白等电点1~2个单位,尽量减少盐或者其他组分(比如甘油)。避开磷酸缓冲液,因为磷酸根容易与钙/镁离子互作,形成盐晶。根据需要是不是需要加还原剂如DTT等。
4. 悬滴法结晶实验的建立,预计消耗时间:2 hr/kit
(1)挑选需要用到的 Crystallization Kits(见附录),按照 Kits条件的顺序,在结晶板(一般 24孔)的盖子上做好编号,每完成一块结晶板的编号,切记将结晶板的盖子与底座做上唯一标记,以防在后续步骤中不同结晶板的盖子与底座混淆。
(2)完成对结晶板的编号与标记后,一一对应地将Kits中各条件溶液加入到各孔中,即加入下槽液,下槽液的体积一般为250~300 μL(如果是优化条件,为方便计算,体积为500 μL。
(3)根据目的蛋白的个数或浓度,预先设计好在一块盖玻片上点几个悬滴,确定数目(如4~6个),进一步确定悬滴间的位置,在结晶板的盖子上最好画一个盖玻片的示意图,将悬滴间的位置关系在示意图上标记清楚,留意盖玻片正反面视角观察导致的悬滴间位置关系的差异,同时在结晶板的盖子上标记清楚姓名、实验日期、蛋白名称、蛋白浓度等相关信息。
(4)按照设定的顺序,将目的蛋白点在盖玻片上,吸取对应的下槽液与之混合(目的蛋白的滴加量根据浓缩后的体积而定,确保目的蛋白的量是足以点完所有条件的,一般目的蛋白的滴加量为0.2 μL;下槽液的滴加量取决于其与目的蛋白的比例,一般为1:1)。
5. 悬滴混合完成后,将盖玻片盖在对应的孔上,压片时用力适中,确保密封(如果结晶板没有自带封胶,那么需要在加入下槽液之前,在每个孔边缘人工均匀地涂上凡士林等封胶);
关键步骤:每点完一块板就随即将其移入18 °C恒温箱中,直至点完所有条件。
6. 不同蛋白结晶速度快慢不一,因此观察晶体形成情况的时间也没有规律,一般是在第二天进行第一次观察,将结晶板从恒温箱中取出,在显微镜下逐孔观察,注意观察时尽量迅速,避免光线、温度等外界条件的变化对晶体造成影响。
7. 根据经验,对每孔中结晶情况进行判断,如遇疑似目的蛋白的晶体,将对应的悬滴标记出来,留待进一步确定或冻存(大多数情况下,初步筛选一无所获是很正常的,少数幸运情况下,会观察到晶体或一些疑似晶体的物质,可能存在各种形态,或太密集,或太小,或极不规则等等,此时就需要对结晶条件进行优化)。
8. 观察结束后,将出现疑似晶体的孔的编号记录下来,随即将结晶板放回恒温箱中,对照Kits的说明书及母液管壁上的注释,将记录下来的编号对应的条件挑选出来,并另记于实验本。
9. 根据目的蛋白初筛时出晶的大概时间,对优化的条件进行显微镜观察,如果能够观察到单个晶体,应该尽快将晶体冻存起来,留待衍射。
影响出晶的试剂成分参考
1. 聚合物分子量大小:
不同分子量的聚合物比如PEG,和水作用方式不同,沉淀蛋白能力也不同。一般来说,分子量大的PEG,沉淀效果更强,更容易产生晶核。相反,分子量小的PEG,更不容易让蛋白沉淀和结晶。因此有的蛋白在较低浓度高分子量PEG(比如PEG8000)下能长晶体,但是不能在高浓度低分子量PEG条件下出晶体。另外,低分子量PEG,包括EG,PEG400等,都是很好的防冻剂。在高分子量PEG条件下长的晶体,可以添加20%左右的EG或者PEG400来防冻。根据PEGs的相似性和不同性质,优化晶体的时候,可以尝试不同分子量的PEG,以获得高质量单晶。
2. 盐的种类和选择:
盐的种类很多,对蛋白质结晶的影响也各有不同。总的来说,基本符合Hofmeister定律。电解质溶液的表面张力表现出“特殊离子效应”,表面张力会随着盐溶液浓度的增加而增大,而在浓度增量相同时不同的电解质溶液的表面能增量不相同,这个现象被认为是Franz Hofmeiste效应。从大量实验看,Hofmeister序列离子对溶液的影响在盐浓度高的溶液中较明显,且阴离子的影响要大于阳离子的影响。模拟研究表明,溶剂化能在离子和周围水分子之间的变化是Hofmeiste效应形成机制的基础。Hofmeister序列如下:SCN-<I-<ClO4-<NO3-<Br-<ClO3-<Cl-<BrO3-<F-<SO42-<K+<Na+<<Li+~Ca2+。序列中头几种离子增加溶剂表面张力,降低非极性盐析分子的溶解度(盐析),加强了疏水作用。而最后几种离子增加非极性盐溶分子的溶解度(盐溶),增加水的有序性,降低了疏水作用。盐析效应通常用于蛋白质纯化,利用硫酸铵沉淀。然而,这些盐也直接与蛋白质相互作用(蛋白质带电且具有强偶极矩),甚至可以特异结合(例如磷酸盐和硫酸盐与RNaseA结合)。具有强烈“盐溶”效应的离子,如i-和SCN-是强变性剂,因为它们在肽基团中盐化,因此与未折叠的蛋白质相互作用比与天然蛋白质相互作用更强,它们将去折叠反应的化学平衡转移到未折叠蛋白上。在一个含有多种类型离子的水溶液中,蛋白变性更加复杂。

针对性建议(TROUBLESHOOTING)
实验前确保蛋白样品比较均一。高速离心去除蛋白沉淀,提高蛋白均一性。
衍射强度
衍射质量
无衍射
多镶嵌晶体Multiple mosaic crystal
弱衍射10埃
镶嵌晶体
衍射3.5-6埃
多晶体
衍射>2.8埃
单晶体
6. 你的修正收敛,与相似分辨率范围内的结构相比,R因子变化不大且合理。Rfree和Rwork之间的差距小于5%。
7. 检查phenix.refine中MolProbity的输出,并修复任何问题。
8. 提交验证服务器或存储服务器。
一般问题
优化方法
目标和约束
非晶体对称性(NCS)
B-因子/ADPs/TLS
孪晶优化
使用R-free
解释结果
氢原子
其他项
引文
一般问题
使用OpenMP支持从源代码编译,它支持并行化在运算结构因子和梯度计算中使用的快速傅里叶变换(FFT)。
指定'nproc'参数(或GUI中的等效控件),它通过启动多个Python作业来并行化各种网格搜索
OpenMP support与Phenix GUI不兼容
“nproc”与Windows不兼容
这些方法彼此不兼容
2. 我将处理器数量设置为8-为什么phenix.refine仍然只使用一个处理器?
phenix.refine已在内部执行各向异性缩放,并对各向异性校正输出map系数,因此预缩放应该是多余的。
因为在计算2mFo-DFc和类似的map系数时,phenix.refine(如REFMAC)默认用F-calc代替缺失的反射,椭球截断将导致消除的高分辨率反射被map中的F-calc替换。这将始终使图谱看起来更好,因为它们更偏向于模型!
即使您选择针对各向异性缩放数据进行优化,也应始终在提交时将原始未修改数据存入PDB。
优化方法
目标和约束
非晶体对称性(NCS)
B-因子/ADPs/ TLS
孪晶优化
使用R-free
解释结果
1. 我通过MR解决了我的结构并且细化了,并且R-free是45%。map很乱,我看到很多差异的密度,但我的分子周围没有。为什么细化不工作?
氢
其他项
引文
5.5.2 结晶条件优化
简介(INSTRUCTION)
利用晶体学方法获取蛋白结构往往需要付出极大的辛苦,因为很多情况下,获得高质量的晶体是很困难的,大多数时候没有晶体或者只有低质量的晶体,例如出现孪晶、晶体太小、晶体
衍射等等。以下是一些常见关于晶体优化等相关内容。
如果只能得到微晶,那么可以通过改变结晶条件:沉淀剂浓度、pH(有时,0.1 pH单位的改变足以影响出晶)、蛋白浓度(提高液滴中蛋白质的比例可以提高蛋白终浓度)、温度(如果需要4度结晶,可在结晶前预冷所有的溶液并冰上操作)、方法(更大的液滴可以形成更大的晶体,因为液滴中含有更多的蛋白质,平衡的速度更慢。也可以尝试坐滴或三明治滴)、蛋白自身。
一、如果无法提高,可以尝试改变蛋白
1. 配体-蛋白复合物。如果你的蛋白质结合配体可制备配体-蛋白复合物,因为配体的结合可能会连接两个子结构域而降低灵活性,将改变蛋白质的表面特性,可能会导致蛋白构象改变。
2. 均质性。非均质的第三四级结构阻碍结晶。 你的蛋白质分解成一个稳定的蛋白水解片段(或者是“自发的”,或者在一个附加的蛋白酶的帮助下)吗?你的蛋白质与同一家族其他蛋白质的同源性是否降低到n-和c-末端?
3. 结构域。你的蛋白质有结构域结构吗?可通过检查Pfam数据库,检查ProDom数据库,Psiblast搜索获悉。
4. 低复杂性区域。有时,点突变可阻止/启动蛋白质结晶。研究不同的物种是获得不影响功能的点突变集合的最简单方法。
5. 脱糖基作用。对于糖基化修饰的蛋白质,松软和不均匀的碳水化合物可能会干扰结晶。可尝试酶促脱糖。
6. 添加剂及洗涤剂。最受欢迎的添加剂有:甘油,(通常用量1%~25%的甘油),可以阻止成核,并可能给你更少,更大的晶体,并具有双重作为冷冻保护剂的优势。乙醇或二氧六环,这些物质会毒化晶体并使晶体停止生长避免成核过多。二价阳离子,如镁。洗涤剂,如β-辛基葡萄糖苷。Hampton有三个添加剂筛选和三个洗涤剂筛选试剂盒。找到合适的添加剂可能与找到蛋白质单一聚合态的条件有关。尝试使用不同的添加剂得到单一聚合态蛋白。
二、晶体衍射相关影响因素:
1. 衍射:衍射取决于晶体大小。因为散射与晶体中的单位胞数成正比。单位晶胞的数目与晶体的体积成正比,因此将立方晶体的所有尺寸加倍,衍射就会达到8倍。相反,晶体中单位晶胞的数量取决于单位晶胞的大小,而单位晶胞的大小又取决于蛋白质的大小。
2. 秩序:散射取决于每个单元的相同程度。 同一性越强,散射越强。盐比蛋白质衍射更好,因为它更有序。
3. 对称性:对于同样大小的晶体,盐的衍射效果比蛋白质的好,因为单位晶胞要小得多。(盐晶衍射的光斑相距较远,在一个小的振荡角范围内,光斑可能会消失。在高分辨率下会有一些强烈的斑点,不会看到低分辨率的斑点。)
4. 晶体的衍射质量可以随以下任何组合而变化:
材料(MATERIALS)
试剂(REAGENTS):目的蛋白、结晶板、凡士林、硅化盖玻片等耗材
器械(EQUIPMENT):恒温培养箱、体视显微镜
实验步骤(PROCEDURE)
1. 确定晶体生长条件。本实验室自己配制的晶体母液,因缺少某些试剂,该试剂可能由其他试剂替代。建议在确定晶体生长条件时以母液管壁上标注的说明为准。
2. 拟定优化方案。一般情况下将沉淀剂浓度拉梯度(当然也有其他方法,下文中会有所涉及),具体的做法是先设定梯度的两端值(即沉淀剂的最小浓度和最大浓度),随即配制好相应的两份母液,然后吸取两份母液按不同比例混合以完成梯度设置(例如,出晶的条件中沉淀剂是2.0 M的硫酸铵,那么可以将梯度设置在 1.5 M 至 2.5 M之间,配制两份结晶母液其含有沉淀剂浓度分别为Solution A(1.5 M)和Solution B(2.5 M),注意这两份结晶母液中只改变了沉淀的浓度而其他成分的浓度和原始结晶条件一致。如果准备点6个孔的话,可以按如下比例分别吸取两份母液混合于相应孔中,如下表:
1 | 2 | 3 | 4 | 5 | 6 | |
Solution A (μL) | 500 | 400 | 300 | 200 | 100 | 0 |
Solution B (μL) | 0 | 100 | 200 | 300 | 400 | 500 |
3. 当然也可以按照每50 μL为基础变量进行递减/递增,这样会产生11个条件。
4. 如果要以pH为优化变量,或者设置条件组合,方法同上述类似。
5. 根据目的蛋白初筛时出晶的大概时间,对优化的条件进行显微镜观察,如果能够观察到单个晶体,应该尽快将晶体冻存起来,留待衍射。
针对性建议(TROUBLESHOOTING)
1. 如何将多块镶嵌晶体制成单块晶体?
一些多晶体可以使用玻璃纤维轻轻分开。
拍摄新鲜的晶体。晶体在生长后的几天内就会变质。
镶嵌晶体需要仔细的数据收集。如果镶嵌性不会导致斑点彼此重叠,则可以进行数据收集。
2. 如何使弱衍射晶体进一步衍射?
注意辐射损伤。有些冻存的晶体在X射线照射下会衰减。晶体死亡表现在第一幅衍射图像上有微弱的衍射,第二幅图像上的衍射更少。
拍摄新鲜的晶体。轻轻地处理晶体,使用大环来冷冻。
脱水。将晶体置于较高的沉淀剂条件下使细胞收缩。这可以在分辨率上产生惊人的提高。
退火。把晶体冷冻在低温流中(不要射击),把它们放回室温,把它们放回结晶溶液中几分钟,然后重新冷冻。
相关文献及书籍:J. M. Harp, D. E. Timm and G. J. Bunick (1998). Macromolecular Crystal Annealing: Overcoming Increased Mosaicity Associated with Cryocrystallography. Acta Cryst. D54, 622 - 628. 以及J. I. Yeh and W. G. J. Hol (1998). A flash-annealing technique to improve diffraction limits and lower mosaicity in crystals of glycerol kinase. Acta Cryst. (1998). D54, 479 – 480.
5.5.3 晶体冻存
简介(INTRODUCTION)
当获得了目的蛋白的晶体后,我们如何让它能够忍受得了X-Ray一定时间内的持续照射,并获得较好的衍射数据呢?
因为晶体直接被暴露在X-Ray的照射下,在极短的时间内,晶体内部的排列顺序就会受到破坏而无法获得足够的衍射数据,因此就需要一定手段来保护晶体。研究发现当晶体处于超低温的环境中,可以延长晶体暴露于X-Ray的时间,维持内部分子的排列顺序。因此,现普遍采用低温冷却的方式来保护晶体以获得更多的衍射数据。然而,直接低温冷却晶体会致使晶体内部产生冰晶,以至于破坏了蛋白晶体内部的有序结构,因此保护晶体的各种防冻剂被科学家们逐渐通过测试而挖掘出来,成为晶体学实验不可或缺的重要一环。防冻剂的选择是需要实验人员根据经验和实际情况多次摸索,也可以凭经验自由组合一些防冻成分已达到实验目的。
常用晶体防冻液
Cryoprotectant | Concentration |
Glycerol | >25% |
Ethylene glycol | >25% |
PEG-400 | >25% |
2R,3R-(-)-Butane-2,3-diol | >25% |
Paraffin | >25% |
2-propanol | >25% |
2-Methyl-2,4-pentandiol (MPD) | >25% |
Glucose | >25% |
Xylose | >25% |
Sucrose | >25% |
Li2SO4 | >2 M |
材料(MATERIALS)
试剂(REAGENTS):防冻剂、结晶母液、Puck、Cryopin、枪头 等耗材
器械(EQUIPMENT):液氮(罐)、绝热盒、显微镜、钳子、移液枪
实验步骤(PROCEDURE)
1. 防冻剂的选择非常关键。虽然上表多种化合物都有防冻效果,但是用法有些不同。首先尽量采用相似原则,也就是说选取与结晶条件相同或者接近的组分。比如如果晶体是在含有PEG条件长的,通过增加PEG的浓度或者添加高浓度小分子量的PEG,达到防冻效果。如果晶体是在高盐条件长的,可以首选具有防冻作用盐,比如锂盐,其次是小分子糖类葡萄糖、木糖、蔗糖等。甘油具有降低电导的作用,往往对在高盐条件下的晶体不利。其次,通过混合多种组分防冻剂,最大限度减少对晶体造成损伤。比如10%甘油,10% PEG,10%蔗糖等。防冻剂的选择还要考虑的防冻剂在结晶条件下的溶解度。比如硫酸锂溶解度较低,很难达到防冻效果。
2. 配制防冻液时,需要保持原条件沉淀剂浓度相同或者适当增加。不能简单在原条件里加防冻剂,这样会造成沉淀剂浓度降低。防冻液的成分是所有成分的最终浓度,配制时需要考虑体积的改变,以最终体积为准。
3. 配制好防冻液,用移液枪稍微吸取一些防冻液于枪头顶部,置于盛有液氮的绝热盒中数秒,移出液面,观察防冻液是否结冰,如结冰(在液氮中变白),需调试防冻液成分或比例,直至防冻液不结冰,在 EP管上标记清楚防冻液成分和实验日期。
4. 准备好具有不同大小fibre loop(0.2 mm, 0.3 mm, 0.4 mm等)的Cryopin,在没有揭开盖玻片之前,在显微镜下参照晶体大小比较并选择出适合的loop。
5. 将Puck打开(注意Puck的序列号),用钳子将其放入盛有液氮的绝热盒中(保证液氮 的量是足够的,整个实验过程中维持绝热盒中液氮的液面高出Puck 1~2 cm),使其充分冷却。
6. 准备好后,揭开相应的盖玻片,反过来放在结晶板的盖子上,置于显微镜下。
7. 用移液枪吸取少量防冻液(1~2 μL)于悬滴上(期间注意此时晶体的状态,是否融化或者沉淀),再吸取少量防冻液于盖玻片空白处(留用于涮loop)。
8. 用合适的Cryopin小心并迅速地挑取晶体,在空白处的防冻液中涮一下,使得晶体周边比较干净(注意观察晶体的状态)。
9. 晶体状态很好的话,迅速将Cryopin放入绝热盒中Puck的相应位置(注意 Puck中孔的编号和顺序),记录该晶体的各种相关信息(蛋白浓度、结晶条件等),Puck序列号以及Puck中孔的编号,切记loop一旦进入液氮液面以下,务必保持其始终处于液面以下。
10. 按上述操作装满一个Puck后,将Puck的盖子用钳子放入液氮中,使其充分冷却,然后将Puck盖好(注意卡槽);。
11. 将Puck移入Puck架上,小心放入液氮罐中,整个过程始终正置Puck,勿将其倒过来以致loop暴露于空气中;
12. 绝热盒中多余的液氮回收到液氮罐中,液氮罐置于实验室阴凉处。
检查Pin金属杆是否弯曲(不是挑晶体的尼龙环),有明显弯曲的一定不要用。
针对性建议(TROUBLESHOOTING)
1. 晶体冻存的关键环节就是配制合适的防冻液和挑取晶体的操作,此步骤一定要谨慎小心,千万不要让好不容易长出的晶体因为准备不充分和操作失误而毁于一旦。
2. 为了更好的重复,对各项信息要详细记录,包括蛋白的批次,纯化的步骤条件,蛋白的浓度、纯度、储存条件等。这一部分工作一定要仔细完成。Puck有空位置的一定做好记录。否则后果很严重。
5.6 晶体衍射及数据收集
简介(INTRODUCTION)
蛋白质三维晶体结构是通过对一系列晶体衍射数据分析处理的结果。作为结构生物学实验室,晶体数据的收集是我们实验室的一项重要工作。
本文将主要讲述两部分,第一部分介绍如何有效规范的记录、整理晶体的相关信息及冻存晶体的操作。第二部分介绍如何使用HKL2000软件初步处理收集的晶体数据。
实验步骤(PROCEDURE)
1. 准备工作
(1)课题申请:进入上海光源收集数据需提前课题申请,填写相关用户信息、实验安全审核等信息方能取得使用资格。每年1月至7月,每月有一次晶体衍射机时。
(2)人员分配:我们实验室每月会派遣人员去上海光源负责晶体衍射实验,目前该任务主要由李越龙同学负责。同时,如果同时有几位同学都准备了冻存的晶体样品,和金老师商量后,有样品的同学可以跟随李越龙一起去上海收集数据。
(3)光源用户注册:根据上海光源用户使用规则,每个进入上海光源进行实验的人员必须提前在上海光源管理系统网站申请个人账号,获得上海光源个人ID号。申请光源个人账号的步骤如下:
(4)登陆上海光源用户课题管理系统http://ssrf.sinap.ac.cn/proposals/default.aspx 进行用户注册在填写个人基本资料、接受安全培训并通过考试测试后取得用户编码(附录有测试答案)。个人需牢记自己的用户名和密码并告知李越龙自己的ID号,方便其记录并安排住宿,否则将无法入住光源宾馆,也无法进入光源收数据。
(5)办理出行证明:提前到生科院找潘主任加盖学院公章,以便携带液氮罐上火车。
2. 冻存、收集晶体
3. 出行指南
(1)科大→合肥南站→上海虹桥火车站:108路(6:00~21:30/22:30):稻香村→合肥南站,出租车约20元。订购动车或高铁火车票,约200元,时长3小时。
(2)虹桥火车站→地铁2号线(05:30~22:45):地铁入口就在火车站内,乘坐方向为浦东国际机场方向,约18站到张江高科站下车,时长约一小时,单程票价6元。
(3)地铁站5号出口→应用物理所(张衡路):地铁站出口跨过马路左边方向有公交站和小吃摊,张南专线/188路或其他的:张江地铁站→3站路到张衡路科苑路下车(离光源近)或2站下车到马路对面吃饭,票价1或2元。也可打车,约12元。返回的路线可以到科伦路做公交,因为张衡路上只有一趟班车。
(4)到光源后,从门卫那儿根据自己的用户编号领取剂量计和用户卡。
(5)宾馆房费200元/天可刷卡,但押金必须交现金,房间12点前要退房。
(6)光源供餐时间:早餐供应时间:周一到周五7:30~9:00;周六到周日8:00~9:00。午餐:11:00~1:00,晚餐:5:00~7:00。注意供餐时间,以便选择时间适时就餐。
关键步骤:做好准备工作:身份证、来回火车票、整钱和零钱、硬盘和数据线、手机和充电器、水杯食品等。可以办理上海市的交通卡,不用每次都备零钱(市内交通,吃饭费用一律不给报销)。
4. 数据收集
(1)开门:先按关闸,再按开门。
(2)放PUCK:将液氮池上的陀螺拿开放在专门的位置,打开盖子(注意电线),旋紧PUCK,放进液氮池,PUCK的凹槽对准定位杆(有两根)。将盖子盖上大概在中间位置,不要盖上陀螺。记录好每个位置对应的PUCK。
(3)关门:按搜索1→按搜索2→人走出来→按关门→按住开闸3秒→开闸灯亮可以走了。
(4)操作演示器:如果是接着别人的机时使用,点击初始化(点击之后会变成另一个名字,再次点击即可)。鼠标点样品→点Mount→点Center(对焦)多旋转几个角度,确保所要收集数据的旋转角度内晶体都处于Focus上→点Collect→命名存储数据路径的文件夹、样品编号、设置参数、收集(0#为单张image用于测试,1#为多张image换样品后要点reset)。
A. Energy不要动,distance表示detector与样品的距离,越近收的数据越多但也可能有过多噪音,因此根据测试分辨率的大小判断用多少距离。Angle表示每拍一次样品旋转的角度,一般为0.5~1°,在shutterless detector比如18U/19U一般是晶体mosaicity 的一半。Expose 表示每次拍照曝光的时间,可根据测试时点数判断,点数过多减少曝光时间,曝光时间太长样品更易损坏。如果光很强可以使用衰减。Image 表示拍多少张,可以根据空间点群判断,但在样品未损坏的前提下可以尽可能多的拍,一般至少360。
B. 衍射图点分散会比较好,如果连成线一般表示栾晶,可以选择边沿的位置曝光通过Beam stop看光的强弱,正在收集的数据如果想停止收集可以通过pause和abort 停止。
关键步骤:一般不要使用衰减,收至少360°,可以一次性把晶体打坏。
(5)记录每个晶体x-ray的参数(距离、能量、角度、命名、曝光存储路径等)。
(6)换样:点下一个样品→Mount→同步骤4)。
(7)最后一个样品收集完成后点dismount→关闸→开门→取出PUCK。但不要关闭可视化软件。
关键步骤:
遇到空loop死机后,点击两次reset→点击initiate两次。
样品收集完毕后不要关闭软件,如果关闭,点击terminal窗口→↑键找回BlueIce命令→Enter→点击Active(Collect页面的下面)。
(8)Copy 数据至自带的U盘。
关键步骤:
线站经常会掉光,听到广播掉光后,到上游的控制箱→旋转钥匙到允许→关闸→下游关闸→旋转钥匙到禁止,重新注入光后,允许→上游开闸→下游开闸。
附:
常见空间群对应的最少收集角度。最优策略下,需要用HKL2000初步处理,根据Strategy 计算出来的起始收集角度。总的收集角度一般是下表最少角度的两倍。
空间群 | 最少收集角度 |
P1 | 360 |
P2 | 180 |
P3 | 120 |
P4 | 90 |
P6 | 60 |
5.7 单晶衍射数据收集要点
简介(INTRODUCTION)
数据收集这一步非常重要。下面是一些需要注意的地方,确保对每一颗晶体,收集到最高质量的数据。
1. 晶体聚焦:非常重要,需要在0度,90度,180度和360度多角度旋转,确定是否在中间。有的时候晶体环太大,收集过程中的抖动,会移位。收集过程需要监控数据。看看衍射降低,是不是由于晶体移动了。这点非常重要!
2. 能量:晶体线站一般有推荐最优波长,比如在1埃左右,12.3 keV,作为收native晶体衍射数据的默认波长。如果是有重原子的晶体,根据需要调整。比如先扫重原子,再确定不同MDA/SAD波长。
3. 光斑大小:光斑不是越大越好,也不是越小越好。一般和晶体差不多大的光斑最好,一般采用50~200微米光斑。光斑大于晶体,容易增加背景。光斑太小,没有把整个晶体利用上,衍射没有到极限。如果晶体太大,比如大于0.5毫米,光斑可以稍小。如果大晶体,且内部不均一,有可能存在晶体的畸形,大光斑收集的衍射点模糊,不能用。这种情况可以用较小光斑,比如5~10微米。
4. 衍射时间和衰减attenuation:需要根据晶体衍射能力和抗辐射能力确定。一般来说,衍射时间越长,衍射越强。X射线能量越强,衍射越强。但是长时间曝光,会增加探测器的背景和晶体衰减。由于晶体的decay是自由基介导的,需要一定时间。因此经验上,用高剂量短时间,比低剂量长时间收到的数据质量更高。因此,首先需要用不是最好的晶体做测试。晶体在收集一整套数据后,观察衍射能力是否明显降低。再选定特定类型晶体能承受的X-射线能量,比如100%或者50%能量。然后确定曝光时间,一般0.2 s~2 s范围。目前17U线站最强,一般不用衰减的话,采用0.2 s至0.5 s。19U能量稍弱,采用0.2 s至1 s。
5. 数据需要收集的角度:也需要根据晶体抗辐射能力和空间群确定。初步处理以后,确定晶体空间群。如果P1,至少收720度;P2至少360度;P3是240度。实际情况是,如果晶体能够承受,时间允许,尽量加倍收。以后处理的时候可以不采用后面的数据。但是万一数据不完整,后悔来不及了。
6. 每张图的角度:有研究发现fine-slicing有好处。结论是mosaicity的一半最优。因此如果时间允许,用较小角度,而不是所有都用1度。19U建议默认0.5度,而17U新检测器非常快,建议用0.1-0.5度。
7. 数据初步处理:建议收集现场处理数据,这样子可以确定所收集的数据质量。万一发现问题,晶体还在,可以重收。并且现场就把每套数据处理完的分辨率、完整性等数据整理纪录,避免时间长后弄错。空间群和收数据的策略有关。因此初步确定空间群后,可以开始解结构。结构解出来,以能修下去为准,且对后面优化收集参数也有指导作用。
8. 数据纪录备份:所有晶体和数据都需要有详细纪录,回实验室后及时做备份。数据需要全部重新理优化,及时解析结构。根据数据质量和结构解析的情况,确定下一步优化晶体的情况。
9. 多少晶体,收多少次数据才够?一颗晶体足够解析结构。原则上,一种晶体(或在同一条件长的)每次不多于8个。事先需要优化,得到最好的晶体。长得单晶,三维最大,没有明显瑕疵。只带最好的晶体去收数据。每种晶体在每次收数据后都要得出一个结论,比如这个条件是否可以,是否需要改进。我们不建议同一个晶体重复收集很多次数据(比如三次)。
10. 数据处理cutoff:你的数据切到哪?需要综合考虑,比较主观。保守的标准是I/σ=2。激进的标准可以是I/σ=1。同时需要参考其他标准,比如completenesss 85%以上,冗余度redundency>5(至少>3),CC(1/2)>66, Rmerge 和Rmeas,不是很重要,可以参考Rpim。
11. 空间群的决定:根据数据建模,systemic absence初步确定,能够解出结构并且把结构修好是金标准。
5.8 XDS预处理衍射数据
简介(INTRODUCTION)
XDS是一个用来处理X-ray晶体衍射图的软件包。包括XDS,它能处理一套数据;XSCALE,用于scaling多套数据;XDSCONV,用于把XDS输出的数据转换成其他格式。同时还有cellparm, 2cbf and merge2cbf. XDS-viewer,后者是一个独立的软件,用于显示衍射图的。更多信息,请参考xdswiki:http://strucbio.biologie.uni-konstanz.de/xdswiki/index.php/Main_Page
目前该软件主要还是在linux/unix系统下以命令的形式运行。
实验步骤(PROCEDURE)
一、使用前安装(以MAC OSX为例):
1. 把下载的安装包解压,把XDS-OSX_64文件夹移到Applications中。
2. 打开terminal,输入:echo $SHELL,查看自己电脑的shell,一般都是bash shell。
然后继续输入:open –a TextEdit ~/.bash_profile,这时会弹出新的窗口,上面显示的都是隐藏命令。在这个新窗口中另起一行,输入:export PATH=full_path_name_to/XDS-OSX_64:$PATH
export KMP_STACKSIZE=8m
3. 在terminal中进入XDS-OSX_64文件夹,输入:pwd,会显示该文件夹的路径。将此路径复制粘贴到第四步中的full_path_name_to/XDS-OSX_64,代替它。回车。
4. 到终端中输入:xds,看是否可以运行。
XDS使用一段时间会提醒过期,这时可以到官网http://xds.mpimf-heidelberg.mpg.de/html_doc/XDS.html 重新下载安装包,按照以上步骤重新安装即可。
二、数据处理时:
1. 修改XDS.INP的参数:
!后面一行表示软件跳过去,也就是不执行的命令。请使用适当的文本编辑器,比如linux 下nedit或者Mac下TextEdit。
2. 在存有数据的文件夹里,新建一个xds的目录,然后进入改目录:mkdir xds/cd xds
3. 把以前的xds文件夹,包括所有输入文件INP文件拷贝过来:Cp *.INP xds/
4. 使用适当的文本编辑器,比如linux下的nedit或者Mac下的TextEdit:Nedit XDS.INP / open –a TextEdit XDS.INP
5. 你将会看到如下内容。红色的地方是需要注意或者修改的地方,其他地方一般不用变。
6. DETECTOR=PILATUS这是18U/19U的shutterless的detector。它有很多块组成。和17U的不同。如果是在17U收集的数据,请另外打开一个针对17U的XDS.INP文件。
下面这些无论在哪里收集,都是一样的。
!====================== JOB CONTROL PARAMETERS
!JOB= XYCORR INIT COLSPOT IDXREF DEFPIX XPLAN INTEGRATE CORRECT
!JOB=ALL
JOB= DEFPIX INTEGRATE CORRECT
这一部分是选择任务。第一次选JOB=ALL。确定空间群后,选择下面的JOB=DEFPIX INTEGRATE CORRECT
!====================== GEOMETRICAL PARAMETERS
!ORGX and ORGY are often close to the image center, i.e. ORGX=NX/2, ORGY=NY/2
ORGX=1224.0 ORGY=1253.5 !Detector origin (pixels). ORGX=NX/2; ORGY=NY/2
DETECTOR_DISTANCE= 400.0 !(mm)
ROTATION_AXIS= -1.0 0.0 0.0
! Optimal choice is 0.5*mosaicity (REFLECTING_RANGE_E.S.D.= mosaicity)
OSCILLATION_RANGE=1 !degrees (>0) 转角
X-RAY_WAVELENGTH=0.9785 !Angstroem 波长
INCIDENT_BEAM_DIRECTION=0.0 0.0 1.0
FRACTION_OF_POLARIZATION=0.99 !default=0.5 for unpolarized beam
POLARIZATION_PLANE_NORMAL= 0.0 1.0 0.0
!======================= CRYSTAL PARAMETERS ==============
!SPACE_GROUP_NUMBER=196 !0 for unknown crystals; cell constants are ignored.如果不知道,就跳过去或者输入0,前面一次输出的可以作为下一次的输入。
! UNIT_CELL_CONSTANTS= 180.7 180.7 180.7 90 90.0 90.0
SPACE_GROUP_NUMBER= 168
UNIT_CELL_CONSTANTS= 123.81 123.81 120.15 90.000 90.000 120.000
FRIEDEL'S_LAW=TRUE ! Default is TRUE.
!REFERENCE_DATA_SET= CK.HKL !Name of a reference data set (optional)
!==================== SELECTION OF DATA IMAGES ==============
!Generic file name and format (optional) of data images
NAME_TEMPLATE_OF_DATA_FRAMES=../TJ21-7_1_00???.cbf !CBF 数据文件路径
DATA_RANGE=1 180 !Numbers of first and last data image collected 使用的数据
BACKGROUND_RANGE=1 6 !Numbers of first and last data image for background
SPOT_RANGE=1 90 !First and last data image number for finding spots,用来初始选点的数据,一般是使用数据的一半。
!==================== DATA COLLECTION STRATEGY (XPLAN) =============
! !!! Warning !!!
! If you processed your data for a crystal with unknown cell constants and
! space group symmetry, XPLAN will report the results for space group P1.
!STARTING_ANGLE= 0.0 STARTING_FRAME=1
!used to define the angular origin about the rotation axis.
!Default: STARTING_ANGLE= 0 at STARTING_FRAME=first data image
!RESOLUTION_SHELLS=10 6 5 4 3 2 1.5 1.3 1.2
!STARTING_ANGLES_OF_SPINDLE_ROTATION= 0 180 10
!TOTAL_SPINDLE_ROTATION_RANGES=30.0 120 15
!====================== INDEXING PARAMETERS ===============
!Never forget to check this, since the default 0 0 0 is almost always correct!
!INDEX_ORIGIN= 0 0 0 ! used by "IDXREF" to add an index offset
!Additional parameters for fine tuning that rarely need to be changed
!INDEX_ERROR=0.05 INDEX_MAGNITUDE=8 INDEX_QUALITY=0.8
SEPMIN=4.0 ! default is 6 for other detectors
CLUSTER_RADIUS=2 ! default is 3 for other detectors
!MAXIMUM_ERROR_OF_SPOT_POSITION=3.0
!MAXIMUM_ERROR_OF_SPINDLE_POSITION=2.0
!MINIMUM_FRACTION_OF_INDEXED_SPOTS=0.5
!======= DECISION CONSTANTS FOR FINDING CRYSTAL SYMMETRY =========
!Decision constants for detection of lattice symmetry (IDXREF, CORRECT)
MAX_CELL_AXIS_ERROR=0.03 ! Maximum relative error in cell axes tolerated
MAX_CELL_ANGLE_ERROR=2.0 ! Maximum cell angle error tolerated
!Decision constants for detection of space group symmetry (CORRECT).
!Resolution range for accepting reflections for space group determination in
!the CORRECT step. It should cover a sufficient number of strong reflections.
TEST_RESOLUTION_RANGE=8.0 4.5
MIN_RFL_Rmeas= 50 ! Minimum #reflections needed for calculation of Rmeas
MAX_FAC_Rmeas=2.0 ! Sets an upper limit for acceptable Rmeas
!================= PARAMETERS CONTROLLING REFINEMENTS =========
!REFINE(IDXREF)=BEAM AXIS ORIENTATION CELL !DISTANCE
!REFINE(INTEGRATE)=!DISTANCE BEAM ORIENTATION CELL !AXIS
!REFINE(CORRECT)=DISTANCE BEAM ORIENTATION CELL AXIS
!================== CRITERIA FOR ACCEPTING REFLECTIONS =============
VALUE_RANGE_FOR_TRUSTED_DETECTOR_PIXELS= 6000 30000 !Used by DEFPIX
!for excluding shaded parts of the detector.
INCLUDE_RESOLUTION_RANGE=50.0 2.05 !Angstroem; used by DEFPIX,INTEGRATE,CORRECT 最后数据的最高分辨率。
!MINIMUM_ZETA=0.05 !Defines width of 'blind region' (XPLAN,INTEGRATE,CORRECT)
WFAC1=0.85 !This controls the number of rejected MISFITS in CORRECT; !a larger value leads to fewer rejections.
这一数值控制rejection比率。1代表不采用rejection,一半在0.8-1.0之间。第一轮不采用rejection。最多不超过10%的rejection,在输出的CORRECT.LP中的最后一个表格中可以算出rejection的比率。
!REJECT_ALIEN=20.0 ! Automatic rejection of very strong reflections
!============== INTEGRATION AND PEAK PROFILE PARAMETERS =========
!Specification of the peak profile parameters below overrides the automatic
!determination from the images
!Suggested values are listed near the end of INTEGRATE.LP这一数值INTEGRATE.LP中后面的表格能找到。上一轮的输出作为下一轮的输入。
BEAM_DIVERGENCE= 0.33031 BEAM_DIVERGENCE_E.S.D.= 0.03303
REFLECTING_RANGE= 1.02947 REFLECTING_RANGE_E.S.D.= 0.14707
对XDS.INP进行修改后,保存。然后执行多任务。在terminal输入小写的命令
>xds_par
如果顺利,会完成。如果有错,会输出错误信息。需要重新对XDS.INP进行修改。
打开三个文件:INTEGRATE.LP,CORRECT.LP,XDS.INP
根据前两者的输出,再放到后者作为下一轮的输入,然后循环,直到REFLECTING_RANGE不变了,差不多10-20个循环。
(1)在XDS.INP中输入正确的空间群和晶胞参数。
(2)将job更改为JOB = DEFPIX INTEGRATE CORRECT
(3)根据CORRECT.LP末尾的表格确定分辨率截断值。
(4)查看CORRECT.LP中的“alien”点列表,将它们放入其中以删除异常值
(5)REMOVE.HKL:awk'/ alien / {if(strtonm($ 5)> 20)print $ 0}'CORRECT.LP >> REMOVE.HKL
(6)将INTEGRATE.LP中的2行复制到XDS.INP的输入参数的建议值
*****为输入参数建议的值*****
BEAM_DIVERGENCE = 0.293 BEAM_DIVERGENCE_E.S.D.= 0.029
REFLECTING_RANGE = 1.473 REFLECTING_RANGE_E.S.D.= 0.210
(a)将GXPARM.XDS复制到XPARM.XDS并重新运行DEFPIX INTEGRATE和CORRECT
(b)作为最后的方法,将WFAC1从默认值1.0降低到0.75,但要注意降低的完整性和增加的R free!
(c)运行“xscale XSCALE.INP”进行扩展作业,并检查日志文件“XSCALE.LP”
(d)注意三个标准:最高分辨率壳层的完整性,I/sigma和CC1/2。
(e)可以忽略“R-meas和R-merge”(它们在高分辨率壳层中可能大于100%)。
(f)编辑“XDSCONV.INP”,将数据格式转换为CCP4,CNS,SHELX。并运行“xdsconv XDSCONV.INP”。现在phenix可以直接把XDS输出的XSCALE.HKL文件转换成mtz文件。
(g)注意“REFLECTING_RANGE”是HKL2000的“Mosaicity”概念。
针对性建议(TROUBLESHOOTING)
1. 有的时候自动处理失败,主要还是需要修改的参数,比如距离,角度,detector类型等没有设好。
2. 偶尔是由于前面的衍射图不好,可以选后面的。
3. 如果redundancy足够,可以尝试扔掉后面部分衍射较差的数据,提高分辨率。还可以适当降低WFAC1。
4. 如果第一轮自动处理,确定空间群,但没有进一步integrate。如果对空间群有信心,不如已经在HKL2000处理了。可以强制进入integrate。
5. XDS选点更多,因此数据质量比HKL2000更好,特别是高分辨率,强度较低的点,能够被采纳。因此可以用HKL2000初步处理,最后用XDS处理。
5.9 XDS-J Version Theta
简介(INTRODUCTION)
XDS-J是程金博2017年为XDS写的一个图形化界面(GUI)。本说明针对版本测试版Theta. 问题意见及建议请反馈至knight3a@mail.ustc.edu.cn。
XDS-J是XDS的图形用户界面。它使得X射线衍射数据的处理更加容易和方便。XDS-J可以使大多数过程自动化,提供统计数据并缩短处理数据所花费的时间。简而言之,XDS-J对于XDS的新老用户来说都是一个很有用的工具。
XDS-J由中国科学技术大学结构免疫学实验室的程金博开发。联系方式:knight3a@mail.ustc.edu.cn
推荐屏幕分辨率1280×960,分辨率过低会导致界面不完全显示(界面上方和右方有滚动条)
系统要求(SOFTWARE REQUIREMENT)
Linux System (tested: CentOS 6.9, Ubuntu 16.04)
XDS (download from http://xds.mpimf-heidelberg.mpg.de/html_doc/downloading.html)
xds-viewer
Python (tested: 2.7, 3.4) with module Tkinter and matplotli
XDS-J (version: open theta, last updated: 2017/07/04)
软件安装(INSTALLATION INSTRUCTIONS)
安装XDS:
下载,解压缩并设置PATH环境变量
安装xds-viewer:
移动到PATH变量中的目录,例如XDS的安装路径
$ wget ftp://turn5.biologie.uni-konstanz.de/pub/xds-viewer-0.6.64bit -O xds-viewer
$ chmod a+x xds-viewer
安装Python:
在Ubuntu下为python2安装模块Tkinter和matplotlib:
$ sudo apt-get install python-tk python-matplotlib
在Ubuntu下为python3安装模块Tkinter和matplotlib:
$ sudo apt-get install python3-tk python3-matplotlib
安装XDS:
解压缩并将执行文件XDS-J复制到PATH变量中的目录
基本使用步骤(PROCEDURE)
1. 打开终端,转到工作路径后,键入命令XDS-J。
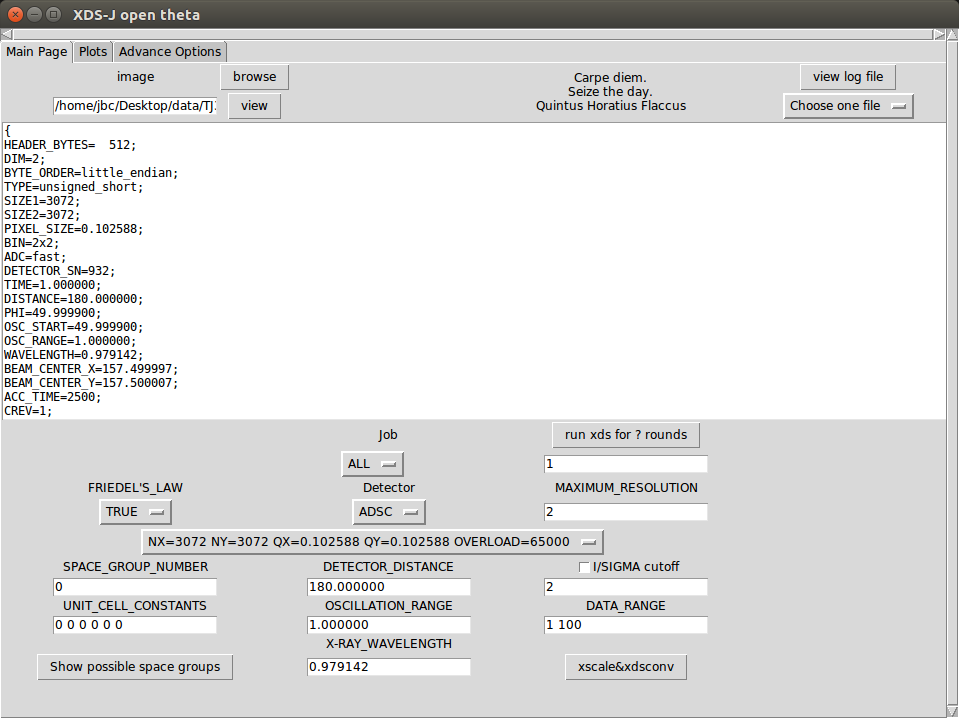
2. 通过image右侧的browse选取一张图片(可以不是第一张)(图片命名方式必须像如下几种这样:ab0001 ab_1_0001 ab_1.0001,可以带格式后缀,可以是压缩文件)(点击view可以观察图片)。
3. 选择探测器类型和参数,若参数不在列表中可以选other然后输入。某些探测器可以自动读取。
4. 输入程序运行所需其它参数或修改自动读取的参数(FRIEDEL'S_LAW在有重原子的时候应为FALSE)。
5. 点击run xds for ? rounds(可以在下面输入打算连续运行的轮数)。若程序正常运行,结束后会显示红字Finished,结果会显示在框中,自动确定的晶体类型和参数自动填入SPACE_GROUP_NUMBER和UNIT_CELL_CONSTANTS的框中。
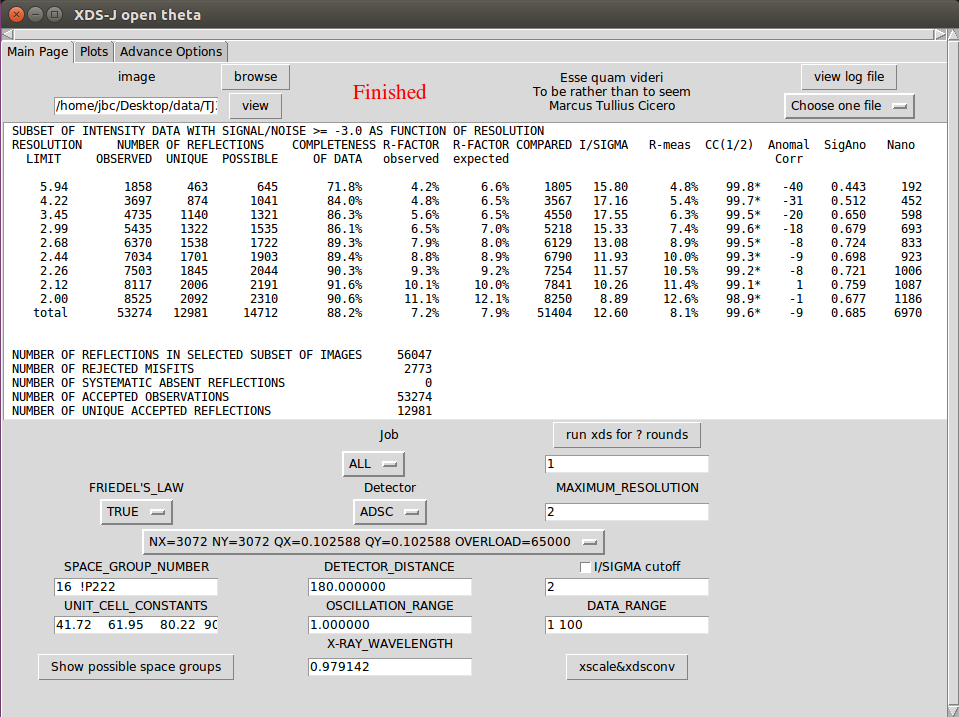
6. 一般Job前两次运行时选“ALL”,之后修改Job为'DEFPIX INTEGRATE CORRECT',根据结果修改最高分辨率(如果在I/SIGMA cutoff前面打勾,运行一次后脚本将根据输入的截断值自动确定下次使用的最高分辨率),也可修改其他参数(如DATA_RANGE,由于辐射损伤有时候要舍弃后面的数据,可参考第二页的图表,或者设置WFAC1 0.85~0.95,EXCLUDE_DATA_RANGE),点击xds再次运行。
7. 多次重复运行XDS以优化结果。
8. 一般最终使最高分辨率对应的I/SIGMA接近2,CC(1/2)大于60,同时总的COMPLETENESS大于90%,NUMBER OF REFLECTIONS中的OBSERVED是UNIQUE的五倍以上,NUMBER OF REJECTED MISFITS小于NUMBER OF REFLECTIONS IN SELECTED SUBSET OF IMAGES的5%(以上数字仅供参考,并不绝对)
9. 点击xscale&xdsconv,在弹出窗口中点击xscale(如果有多套数据,可在此步把它们合并)。再点击xdsconv,最终生成CCP4,CNS,SHELX可用的mtz文件。

其它功能(OTHER FUNCTIONS)
1. 在界面右上角可以选择一个日志文件,然后点击view log file按钮打开查看。
2. 点击Generate XDS.INP only按钮将只生成XDS.INP文件而不运行xds。
3. 点击View XDS.INP按钮可查看XDS.INP文件,更改内容后点击save可以保存。
4. 如果想利用已有的XDS.INP文件运行则点击Run with existing XDS.INP按钮。
5. SPACE_GROUP_NUMBER为0时运行完xds点击Show all possible space group按钮可显示CORRECT.LP文件中判断的所有可能的空间群及参数。
注意
(1)XDS在积分时效果优于其他软件,但有时自动确定的空间群不准确。这时可以先用其他软件(例如HKL-2000,iMOSFLM)确定空间群,再用得到的参数运行XDS。
(2)运行XDS时整个界面将暂时处于锁定状态,不要对其进行任何操作,以免造成未知后果。
(3)程序运行时将产生很多文件,最好在一个单独目录下运行。
(4)显示结果的最大分辨率与输入的最大分辨率不一致可能是因为改分辨率下没有点。
(5)如果结果显示各个分辨率范围的信噪比都很低且差别较小,有可能是晶体位置(ORGX,ORGY)偏离较大或空间群选择错误。
(6)运行失败可能的原因包括:数据本身质量不好(可通过view按钮查看图片),旋转轴的正负错误,最高分辨率过低(例如0),空间群选择错误(改变DATA_RANGE有可能导致自动确定的空间群不同)。
6. 提高信噪比方法:WAC1调低(如0.8,0.9),改变DATA_RANGE,多运行几轮XDS。
7. 探测器类型和参数未知的可以尝试通过读取图片的文件头中的信息再加上用xds-viewer打开确定像素,再参考XDS网站得到。有关参数可在XDS网站(http://xds.mpimf-heidelberg.mpg.de/html_doc/detectors.html)上找。
参数的意义
(详细介绍参见http://xds.mpimf-heidelberg.mpg.de/html_doc/xds_parameters.html)
1. Job为All时将运行XYCORR,INIT,COLSPOT,IDXREF,DEFPIX,XPLAN,INTEGRATE,CORRECT
2. MAXIMUM_NUMBER_OF_PROCESSORS为要使用的处理器数,最大32。
3. DETECTOR,NX,NY,QX,QY,OVERLOAD为探测器的参数。
4. ORGX,ORGY为晶体所在位置,默认为NX,NY的一半,特殊情况晶体偏离中心可通过观察图片后输入。
5. RESOLUTION填最大分辨率(单位A)。
6. NAME_TEMPLATE_OF_DATA_FRAMES为图片所在路径,注意不能有空格,例如/test/TJ_1_???.img以表示TJ_1_001.img,TJ_1_002.img,TJ_1_003.img等。
7. DATA_RANGE为要使用的图片范围,填两个整数,以空格隔开。
8. ROTATION_AXIS一般为1 0 0(但18u,19u是-1 0 0)。
10. SPACE_GROUP_NUMBER第一次运行不知道的话填0,此时UNIT_CELL_CONSTANTS可以空着,XDS可以自动确定空间群,不过有可能不正确。
11. TRUSTED_REGION如果是0到1相当于探测器的内接圆,0到1.414相当于探测器的外切圆。
12. WFAC1值越小,拒绝的点越多,一般在0.8~1.0之间。
13. FRIEDEL'S_LAW在有重离子的时候应为FALSE。
14. EXCLUDE_DATA_RANGE是要排除的数据范围,两个整数用空格隔开,如要排除单张则两个相同数字,该参数可以有多行。
脚本运行原理
1. 通过image右侧的browse选取一张图片会显示图片的头几行,脚本自动确定NAME_TEMPLATE_OF_DATA_FRAMES,DATA_RANGE为所有数据,并从中读取DETECTOR_DISTANCE,OSCILLATION_RANGE,X-RAY_WAVELENGTH等参数(目前只支持ADSC,PILATUS,CCDCHESS三种探测器)。
2. 点击view将调用xds-viewer打开选取的图片。
3. 点击run xds for ? rounds,脚本将自动生成XDS.INP并运行xds_par。若程序正常运行,结束后会显示红字Finished,CORRECT.LP中的结果会显示在右侧的框,获得的晶体类型和参数自动填入SPACE_GROUP_NUMBER和UNIT_CELL_CONSTANTS的框中。
4. 如果在I/SIGMA cutoff前面打勾,下次运行后脚本将根据输入的截断值通过线性插值法估计下次使用的最高分辨率。
5. 点击run xds for ? rounds再次运行时脚本会自动读取INTEGRATE.LP中REFLECTING_RANGE,REFLECTING_RANGE_E.S.D.,BEAM_DIVERGENCE,BEAM_DIVERGENCE_E.S.D.的值加入XDS.INP文件中,并用GXPARM.XDS替换XPARM.XDS。
6. 点击xscale时,脚本自动生成XSCALE.INP并运行xscale。点击xdsconv时,脚本自动生成XDSCONV.INP并运行xdsconv,f2mtz。
5.10 分子置换法解相位
简介(INTRODUCTION)
分子置换(MR)是最常用的解决相位问题的方法。一般要求目标蛋白A有同源蛋白结构B,同源性30%以上。
实验步骤(PROCEDURE)
(1)先用CCP4中的matthews判断一下一个晶胞中有多少个分子。在CCP4 MR 模块下,打开cell content analysis。 这个是晶体中蛋白聚集体数的分析,通过分析晶体含水量得到一个晶胞内的蛋白分子数。需要输入mtz文件,单体蛋白的分子量或者氨基酸数目。由于大多数生物大分子晶体含水量在40%~60%之间,同时最小对称单元中,蛋白数值对应n是整数。可以初步确定n值。这个聚集体数n会在mr中使用,用来更加准备估计含水量作为背景。一般分辨率在2埃的晶体其含水量在50%左右。也可以根据分辨率,指导n值的确定。
(2)步骤:输入mtz文件,再在下拉菜单中选择一种确定分子量的方法。可直接输入(默认)或用残基数,序列文件,pdb文件估计等。点击Run Now。运行完成后得到不同分子数(Nmol/asym)对应的可能性(P(tot))。一般会有一个数字对应的可能性非常大,即一个晶胞中的分子数。
(3)model 选取:进行分子置换的model为已知的同源蛋白结构或硒代得到的pdb,对model的要求是越接近球形越好。一般用单体。从pdb库中下载了pdb后可以用vim编辑,选取自己想要的那一段做model。
(4)Phaser MR做分子置换。该程序在CCP4和Phenix中都有。
(5)使用CCP4步骤:
所有必须项都用深黄色标示出来了。在Define Data栏中选取mtz文件,如果不确定空间群可以把Run Phaser with后的下拉菜单选成all alternative space groups。在Define ensembles栏中选取pdb文件。在Define composition of the asymmetric unit栏中选取序列文件(可用输入分子量或输入残基数代替),Number in asymmetric unit后面输入之前得到的晶胞中分子数。点击Run Now。
(6)使用Phenix步骤:
在Input and general options页中选取mtz文件。在Ensembles页中点击Add file选取pdb文件。在ASU contents页中选取序列文件(可用输入分子量或输入残基数代替)。在Search procedure页中点击Component 1后的按钮Add ensemble中选之前添加的那个pdb文件,Copies to search for填之前得到的晶胞中分子数,如果不确定空间群可以把Also try alternative space group选成All possible in same pointgroup。点击Run。
(7)成功运行会输出一个mtz文件和一个pdb文件。
关键步骤:
如何判断得到的解是否正确。TFZ大于8表示几乎可以确定是正确的解,7~8表示很有可能,6~7表示有可能,5~6表示不大可能,小于5表示不可能。PAK理想情况为0。
如果成功找到解,接下来就可以进行结构修正。
可使用CCP4中的refmac5或Phenix中的phinex.refine。把之前得到的mtz文件和pdb文件作为输入,运行成功会输出一个mtz文件和一个pdb文件。运行前后Rfree,R-factor的值应该有所减少。接下来用COOT同时载入刚才输出的两个文件,手工进行结构修正。然后再用程序进行修正,不断重复这一步。见后面Coot指南。
如果提示mtz文件没有Rfree标签,打开CCP4中的truncate。选取该mtz文件,选中Ensure unique data & add FreeR…。点击Run Now。输出文件就有了。
5.11 结构优化检查清单
简介(INTRODUCTION)
结构修正永远不会完成,但是当人们再也无法改进时,就不用再修正了。以下是我的检查清单,您无需按照订单操作。

实验步骤(PROCEDURE)
1. 确保模型的序列是蛋白质的实际序列。使用Phenix或其他工具提取最终模型的序列,并与您的实际蛋白质序列进行序列比对(根据您的测序结果)。
2. 在coot中检查map和模型。
(1)手动检查2Fo-Fc图谱中5 sigma以上的峰值。
(2)没有未建模的斑点。
(3)没有ramachandran plot outliers。尽力改善它。
(4)检查并修复大多数侧链outliers。
(5)仔细检查结构中的任何配体。
(6)检查水。去除那些密度很弱,即小于11 sigma峰值的那些,以及那些与其他原子距离太近(小于2.2 Å)或太远(大于3.5 Å)的水。
3. 在Phenix.refine中,使用TLS修正。几乎在任何分辨率下都很好。
4. 在phenix.refine中,在修正中使用氢可以改善冲突。只有在非常高的分辨率下(即高于1.5Å)使用氢,否则使用riding模型。
5. 手动或自动优化X射线/立体化学,ADP重量。
5.12 如何修正低分辨率的结构(5-6 Å)
简介(INTRODUCTION)
不同分辨率,结构的修正难度差别很大。分辨率低预示着来自数据的信息少,数据比参数的比值低,结构不确定性大。而最基本的,无论怎么修正,结构的所有键长键角等数值不能违背理论值。
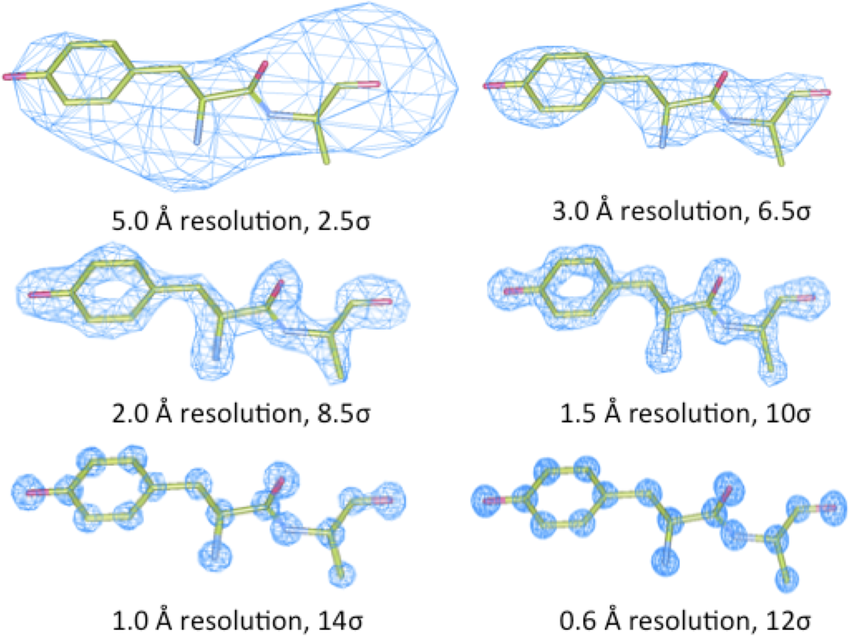
教程 (TUTORIAL)
人们经常使用Phenix.refine来修正低分辨率结构,因为phenix.refine实际上是一个很好的程序,可以针对低分辨率结构进行细化。如何进行取决于您希望完成细化的内容。如果结构已经近似正确,没有重大的构象变化,这里有一些基本的指导原则:
1. 使用紧束缚。权重自动优化在此分辨率范围内不能很好地工作,因此可以同时将wxc_scale和wxu_scale设置为非常低的值(它控制X射线项相对于几何和B因子约束的权重) 是一个很好的起点。建议尝试wxc_scale = 0.025和wxu_scale = 0.05的值。
2. 如果ASU中有多个分子,请使用NCS限制(torsion限制通常是足够的,并且更容易使用,但也可能值得尝试cartesian限制)。
3. 如果你有一个好的高分辨率结构,可以使用参考模型(这可能只是结构的一部分)。
4. 真实空间的修正可能不会起作用。
5. 首先尝试使用非常保守的修正策略,例如刚体和B族因子簇修正。后者可能效果更好,因为增加了对邻近基团的限制。您还可以尝试优化结构域TLS。我自己的经验是,individual coordinate和 B-factor refinement的改进效果最好。后者有点争议,但是在严格限制的情况下,它最终与修正整体B因子没有太大区别。在修正开始时将所有B因子重置为20有助于消除起始模型的偏差。
6. 在此分辨率下应该对于2mFo-DFc电子云图非常谨慎,它看起来几乎就像你的模型。因此需要制作大量的Omit电子云图,如果你有任何重的散射体(金属等),这些将非常有助于确认你结构的正确性。如果你真的需要进行大量的重建,那就更难了,DEN或Rosetta修正可能会有所帮助。
7. 定义二级结构并在结构修正中使用它。如果不使用二级结构限制,那么在起始模型中即便是理想的二级结构在如此低分辨率的修正之后也会恶化。最重要的是,确保您定义的二级结构限制实际上用于修正。您可以使用phenix.secondary_structure_restraints让Phenix自动识别二级结构。至关重要的是,您需要手动查看它,并在必要时进行编辑(最有可能的情况),以获得最准确的注释!
8. 旋转NCS在大多数情况下是比较好的选择,然后在非常低的分辨率下,笛卡尔NCS可能是更好的选择。因此,正如纳特指出的那样,尝试两者并看看哪一种效果最好。
9. 如果真实空间修正表现不佳,请报告错误,因为它将消除旋转异常值rotamer outliers。
10. 如果有Ramachandran outlier拉氏图异常值:手动固定它们,然后使用Ramachandran plot restraints来防止它们再次发生(重要:不要使用拉氏图限制来修复异常值!)。
11. 如果数据集严重不完整,请查看两个2mFo-DFc地图:常规和缺失Fobs“已填充”(phenix.refine输出两个地图)。
5.13 结构解析软件的使用
5.13.1 HKL2000 基本操作
简介(INTRODUCTION)
这部分简要介绍如何使用HKL2000软件处理蛋白晶体衍射数据。由于篇幅的限制,这里很少涉及每个参数的具体意义。
实验步骤(PROCEDURE)
1. 打开“HKL2000”软件并加载衍射数据
(1)点击鼠标右键,在菜单中选择“open terminal”或直接点击终端窗口打开终端窗口。在终端窗口输入“HKL2000”→Enter。此时就打开了HKL2000程序。
(2)打开HKL2000程序之后会自动出现一个对话框,让你从菜单中选择检测器。每个线站的检测器类型在下表中列出,选过之后点击“OK”按钮。

线站 | 17U1 | 18U1 | 19U1 | 19U2 |
Detector | Eiger X 16 M | Pilatus6m | Pilatus6m | Pilatus1m |
衍射仪 | MD2 | MD2 | MD2 |
(3)此时HKL2000的主窗口会打开。进入“DATA”页面,在左侧的“directory tree”中找到你要处理数据所在的文件夹(你的衍射图片不会显示出来,并不是你的文件夹是空的)→点击“Create Directory”→在新出现窗口的输入面板后加上“/hkl”→点击“create”(创建一个名为“hkl”的子文件夹,用于存放处理产生的新文件),将你的衍射数据所在的文件夹设置为“Raw DATA Dir”、新创建的“hkl”文件夹设置为“Output DATA” (不需要人工输入,点击相应的文件夹之后再点击![]() 即可)
即可)
(4)Figure 6 是将子文件夹命名为“proc1”
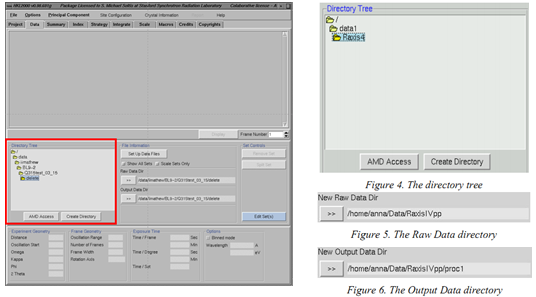
(5)点击“Load Data Sets”选择你要处理的数据(会变蓝),点击“OK”
会出现一个新窗口,如下图。如果跳出来一个新窗口点击“continue”

备注:figure 7 的“Scale Sets Only”是只显示index和integrate之后的文件,所以对于只需要scale处理的数据可以勾选。
(6)如果你的数据有些衍射不好或其他原因需要丢弃,可以点击set controls中的“splict set”将不要的数据抛弃掉。

2. Index和refine
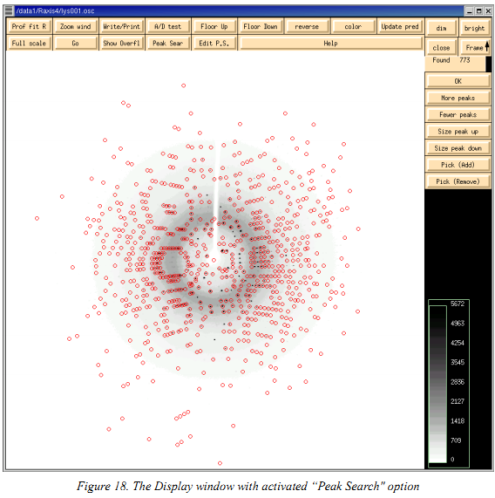
(1)进入Index 界面,填写分辨率,将3Dwindow 填写为“5”、display Frame Number一定要填写为“1”。点击“display”之后会出现一个display窗口。点击index界面的 “Peak search”按钮。用鼠标中键点击display窗口上的“Frame”5次,然后点击“OK”。
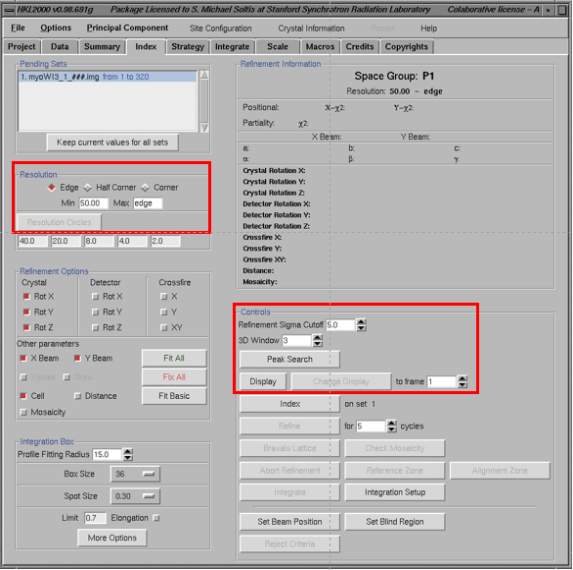

备注:分辨率怎么设置:如果有冰环存在的话就设置为4A。
3D window怎么设置:3Dwindow的数值乘以单张震荡角角度至少是优化后镶嵌度的两倍,对蛋白晶体,5是一个比较合理的数值
(2)点击index窗口的“index”按钮以启动index程序→点击“Bravais table”出现Bravais table窗口,选择“primitive triclinic ”晶格→点击“apply & close”。在display 窗口确认绿色的预测环与衍射点要重合的比较好


(3)Index之后,点击“refine”按钮,refine第一轮之后将refinement options选为“Fit All”再次点击“refine”按钮。注意观察X-X2、Y-X2的值(最好不要超过2,如果绿色的预测环与衍射点重合的比较好的话也可以)→再次点击“bravais lattice table”选择合适的晶格(绿色的)→apply &close→refine。建议从低对称性往高对称性挨个往上走,这是多个refine的循环。

备注:如果预测环与衍射点不符合的话,可能暗示你的晶格类型选错了
X2是衡量预测环与衍射点拟合误差的参数,但是这个数值对于index和后面的integrate影响不大。
3. 设置分辨率、Fitting Radius 和 Spot Size
(1)如何设置spot size
选择太小的spot size,则有些衍射点会被丢失掉,太大的spot size会使衍射点重叠,而重叠的衍射点在scale时会被reject且在计算衍射点的intensity时会将临近衍射点的强度也计算进去。
由于衍射点本身的大小就不统一,所以就要做一个权衡,一般的经验法则是尝试调整spot形状以适应图像上的某些中等的点到强点。
(2)如何设置分辨率
一般来讲,要将分辨率设置为比你看到的稍微好一点,这样即使你设置的不合理还可以在scale时改正过来。相反,如果分辨率设置的保守了,你将不得不在scale之后返回来重新integrate,因为scale并不能使用超过你现在设定的分辨率。确定好分辨率之后,你需要再返回index窗口重新填写分辨率。然后再次refine。这同样是个反复的过程。
(3)如何设置Fitting Radius
Profile Fitting Radius is used to calculate the average spot profile,Generally, the radius is set so those spots on roughly 3~5% of the area of the detector are included in the averaging
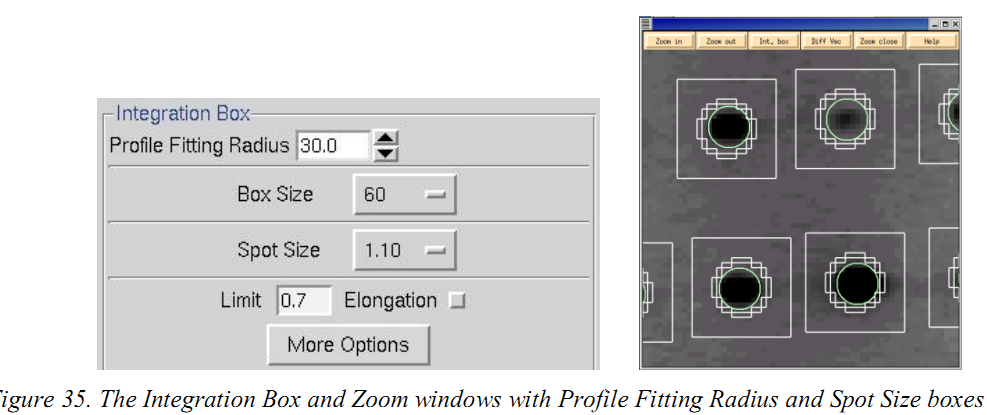
(4)Elongation Limit是什么?
这个选项是考虑到由X射线与探测器在高角度交点引起的斑点的径向伸长。 通过点击 “Integration Box”面板中的“Elongation Limit”按钮激活此功能。 Elongation Limit的默认值(表示为spot伸长率的分数)是0.7。范围从0到1。
4. 3D Window 和 镶嵌度
(1)什么是3D Window
3D窗口是同时进行integrate的连续image的数量。该功能的一个主要优点是将image合并在一起可以在refine中包括更多的衍射点,从而提高index的准确性。
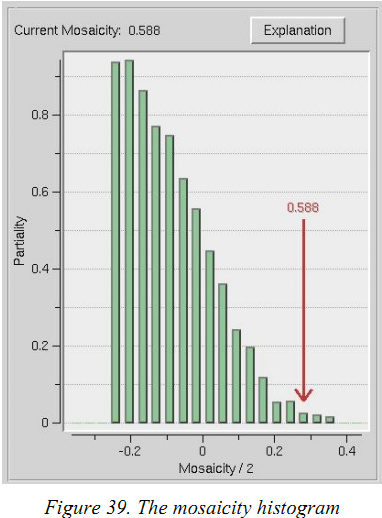

(2)如何设置镶嵌度
设置镶嵌度的最佳方法是使用3Dwindow,在integrate衍射点时对镶嵌度进行refine。正确设置镶嵌度并不总会导致预测环覆盖image上的所有衍射点。特别是,非常强的low order的衍射点可能需要相当大的镶嵌度值才能被预测环覆盖。这些衍射点可能不代表晶体内在镶嵌扩散,可能是由于该晶格点的特别强烈的衍射产生的。你点击“Crystal Information”按钮并输入“镶嵌度”,经验上讲镶嵌度设置得高比设置得低好,但设置的太高可能会导致大量的overlap。
5. Strategy and Simulation(不属于数据处理过程)
(1)Strategy and Simulation的目的是用来决策什么样的参数(曝光时间、距离、震荡角、收集度数)可以得到比较好的晶体数据。
(2)怎么开始策略?
正常Index之后,在确定正确的晶格、适当的分辨率、选fit all refine后,先别开始intergrate。
一旦index之后,进入strategy界面(图41),第一个表就是strategy,点击strategy左侧的“strategy”按钮→出现含很多彩线的图(横坐标omega (Kappa, Chi, Phi)start就是开始角度,,纵坐标delta Omega是总度数)。

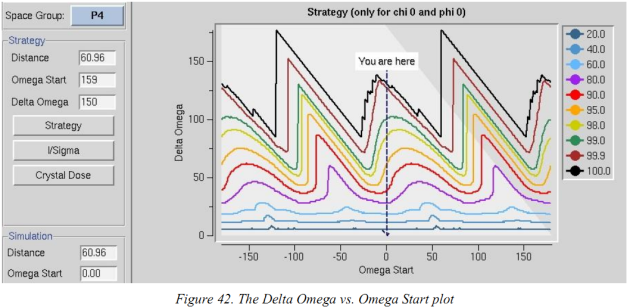
所以Delta Omega vs. Omega(Kappa, Chi, Phi) Start描述的是在某一给定的起始角度要收集相应百分比的数据(彩色线)所需的最少度数(灰色三角部分是由于硬件限制不能收集的区域)。将鼠标移动在窗口中左侧显示出Omega Start and Delta Omega。所以黑色线的最低端的Delta Omega是收集100%数据所需的最少度数,但要从相应位置开始。建议收集360(对称性越高所需度数越少)→点击鼠标,相应的位置的Delta Omega 、Omega Start会填入Simulation面板中→点击“simulate run”出现一个新窗口

用于预测数据收集过程中overlap衍射点的数目。当晶体具有高镶嵌性或长晶胞轴的时候这很重要,这两种情况使部分image中的衍射点出现严重重叠而无法使用(当长晶轴平行于光束时会overlap)。
你可以改图左侧的frame width的值,然后重新“simulation run”(减少frame width的值overlap应该也减少,因为总的点少了),想增加完全记录的衍射点的百分比则增加震荡角。对于overlap比较高的情况,也可以增加距离然后再次SIMULATE RUN,衍射点会分的更开但最高分辨率会低。
上面的四个参数(开始角度、总角度、距离、震荡角)确定之后,就可以用这些参数来收数据了。
6. Integration and scale
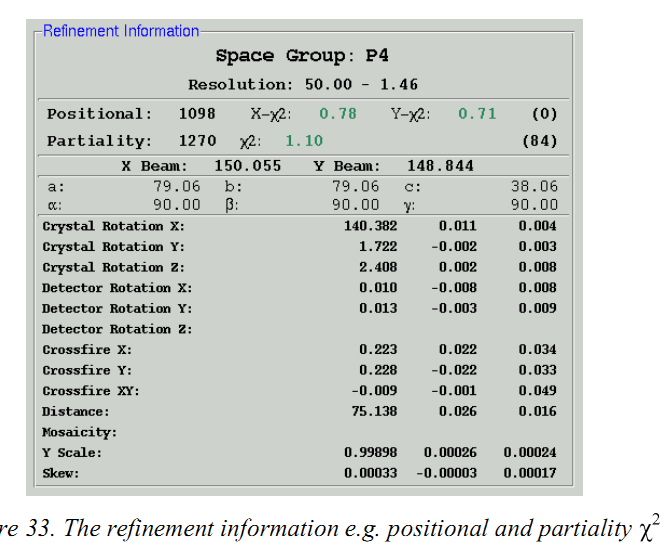
(1)点击index界面的integration按钮, X2,Cell Constants,Crystal Rotations,Mosaicity和Distance vs。 Frame图表的线都应该是水平的,X2vs. Frame 的两条线都应该在1~2之间。

(2)Integration按钮是红的或橙色说明什么
X2值太大,高镶嵌性等导致的,可以尝试re-index。尤其是当晶胞参数变化大时,可以点击“fix mosaicity”这是因为当多晶存在时程序不知道选哪些,但限制了镶嵌性就限制了点。或者就是点阵选错了。
7. Scaling
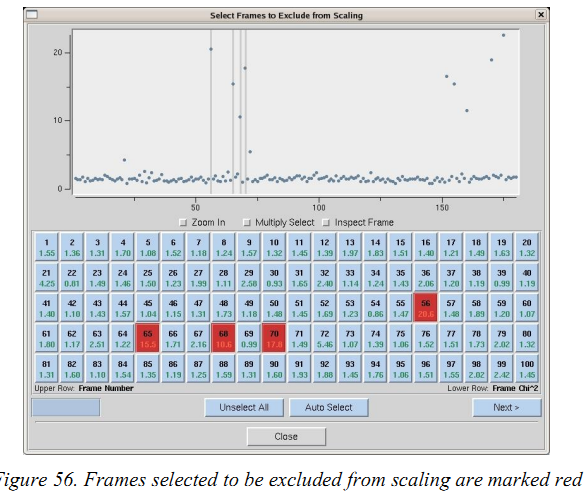
(1)在integrate之后,某些image可能在integrate信息图中具有非常高的离散值。 您可以通过单击EXCLUDE FRAMES按钮从scale中去除这些image。 将出现一个包含image列表的表格。 通过点击image的框或者通过点击“Multiple Select”选择一定范围的image(一次点开头,一次点结尾)。 红色标记的image将被去除。然后关闭表格并重新scale。
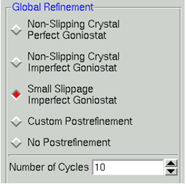
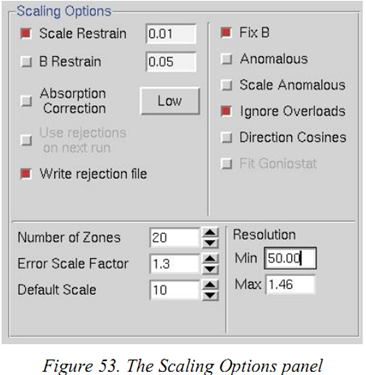
(2)进入“Scaling”界面。选择合适的“Global Refinement”选项(5个选项,最好选择Small Slippage Imperfect Goniostat),填写scaling options里的值
Scaling options 选项框的默认值:Scale Restrain 0.01,B restrain 0.1,选Write Rejection File,选Ignore Overloads,Number of Zones 10,Error Scale Factor 1.3,Default Scale 10、分辨率限制是从.X file读出来的(index和integration之后的值),最好选上“Absorption correction”。
(3)先选primary空间群(布拉维点阵的第一个空间群), 点击“scale sets”开始第一轮scaling,注意X2的统计数据。
(4)第一轮scale之后,检查输出图表并调整“error scale factor”(使X2到1)、然后点击delete reject file,再次点击scale sets。重复这些步骤直到整体X2可接受,然后通过选上“Use rejections on Next Run”继续后继的scaling,再次scale。重复scaling和rejecting的循环,直到新拒绝的数量已经下降到只有几个。 此时scale就完成了。选过“Use rejections in next run”之后根据2σ调整分辨率

(5)下一轮的scale填入的最高分辨率要比信噪比大于2的稍好一点。比如:最初分辨率为50-2.2,第一轮scale后,发现I/sigma=2在2.3A处。所以下一轮分辨率为50-2.3.
(6)现在尝试更高的对称空间群(下一个)并重复scale,保持其他所有内容相同。如果X2大致不变,你可以继续。如果X2更糟糕,那么这是错误的空间群,上一个选择是你的空间群。但有个例外,对于primitive Hexagonal点阵,在P3121 and P3112失败后要尝试P61
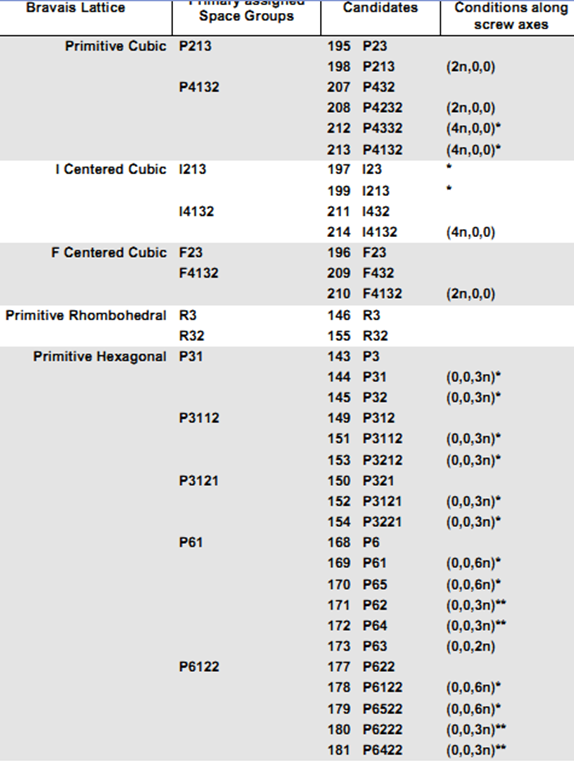

注意:标星号的是相似的空间群,衍射强度的scaling和merge不能解决你的晶体属于哪个空间群。
(7)打开log.file的最下端(点show log file)或模拟的倒易点阵图(点RECIPROCAL LATTICE)检查系统消光。如果空间群是对的,则系统消光的衍射点应该不存在(或很小)。将该列表与其他候选空间群的衍射点条件列表对比,如果你的数据中消光的部分和哪个空间群消光特征一样则这就是你的空间群。
(8)如果证明你的空间群是orthorhombic(正交)的且包含一个或两个旋转轴,你需要reindex使旋转轴与标准定义一样。如果只有一个螺旋轴,你的空间群就是P2221(螺旋轴和c平行),如果有两个螺旋轴,你的空间群是P21212(螺旋轴along a和b)如果index的与这不符重新index吧(点击reindex)。
(9)Scale的输出文件
output.sca:包含scaled reflections(衍射点),晶格单元、space group和h、k、l、I、σ。可以重命名,但最好保留后缀“.sca”

scale.log:scaling 操作的log(记录)文件。可以重命名,但最好保留后缀“.log”
Scale.log是scalepack的log(记录)文件,包含所有读入的“.x”文件(index后的文件)每个scaling循环的记录。可以用来检查scaling的质量。
每个循环之后都应该检查log文件,尤其是:errors、X2和两个R因子。在接近最后有“redundancy”
备注:
什么是Error Scale Factor以及怎么调整它?
ERROR SCALE FACTOR是一个单一的乘法因子,用于输入的。.它应该调整以使X2的值接近1。.默认情况下使用输入的误差(ERROR SCALE FACTOR = 1.3)。 默认值可以向上调如果X2大于1。
Error Scale Factor的合理值是什么?
合理的值介于1和2之间。默认值为1.3。 如果你需要使用远大于2的值,那么就有问题了。
在scale后显示的图表中应该寻找什么?
Scale结束后会有10个chart,每个chart都有说明按钮。有些chart有两个垂直轴,对应的线对应相应的轴,有些chart可以在线性和log图间转换。下面是几个举例:
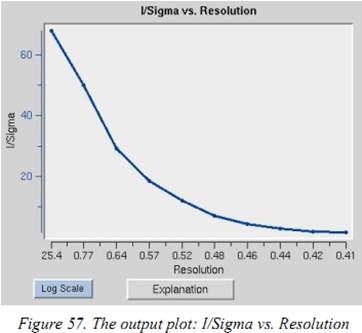
I/sigma vs resolution:I/sigma是intensity与误差的比值,在scale中应该取I/sigma为2的分辨率
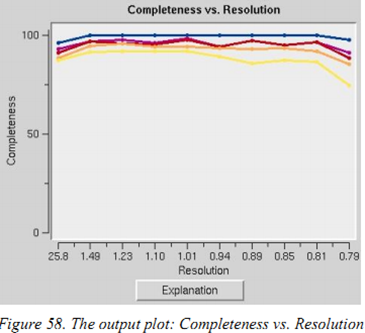
Completeness vs.Resolution:不同分辨率下数据的完整性,蓝线代表全部的衍射点、紫色的是I/σ大于3的衍射点、红色是大于5的、黄色是大于20等等。这一步可以判断你的收集策略是否有效,是否需要更多的数据。
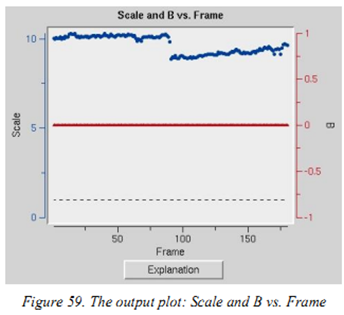
Scale and B vs. Frame:看images之间intensity和B因子是否有大的变化,用于判断晶体是否旋转出束或者受到辐射损害。
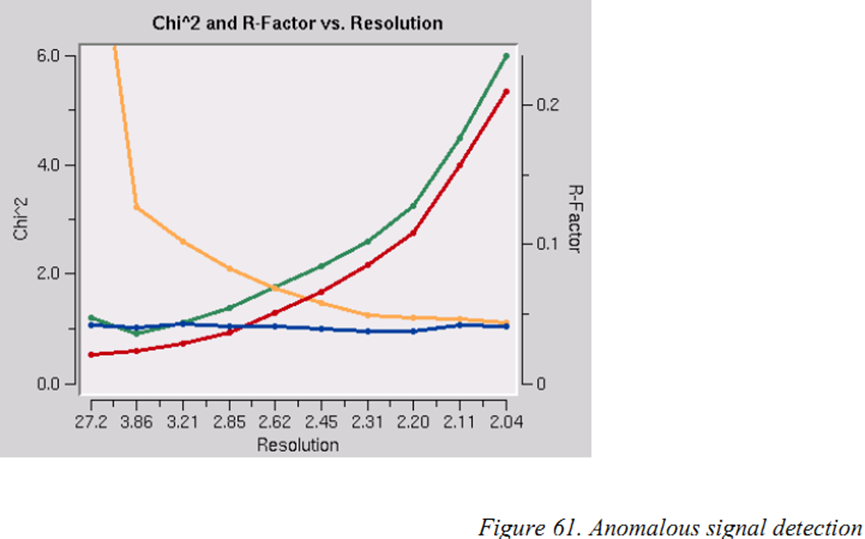
怎么处理含有反常信号的数据
检测是否含有反常信号非常容易:选择“anomalous”选项(不是SCALE ANOMALOUS选项)后数据进行正常的scale和postrefine。这会输出I+和I-衍射点分开的.sca文件。Scale会将独立测量I+和I-数据,两者合并的统计数据反映出I+衍射点和I-衍射点的差异。对于中心部分的衍射点没有I-,所以合并统计只会反应非中心的衍射点。你可以用于计算合并统计的数据百分比通过检查日志文件末尾附近的redundancy表格。在redundancy> 2的列,你将找到百分之多少的数据用于比较。因为你只有I+和I-,所以redundancy永远不会超过2。
通过检查Chi^2 and R-Factor vs. Resolution中的X2的值来检测异常信号的存在。
两条X2曲线显示了分别为合并的(橙色)和未合并的(蓝色)Friedel配对计算的不同分辨率下X2的值。假设errors scale factors是合理的,并且数据中没有有用的反常信号,那么当使用合并或未合并的Friedel配对进行scale时,两条曲线显示的X2对分辨率函数应该是平坦的且平均值约为1。另一方面,如果X2> 1且对于使用合并的Friedel配对进行的scale,X2具有清晰的分辨率依赖性(即随着分辨率降低X2增加),则强烈表明存在反常信号。分辨率依赖性允许您确定在何处切断分辨率以计算具有最佳信噪比的反常差异帕特森map。但请注意,该分析假定错误模型为合理的且当反常信号不存在时X2接近1。
5.13.2 CCP4基本操作
简介(INTRODUCTION)
CCP4英文全称为Collaborative Computational Project,Number 4。CCP4i是CCP4图形用户界面。
CCP4是晶体学软件开发方面的合作,以分配一套晶体学程序和软件库而闻名(Collaborative Computational Project,1994年第4期)。软件库支持一些标准的晶体文件格式,并提供用于命令解析,对称处理和其他常用功能的工具。主要用于X-ray衍射数据的处理,结构解析及分析。
操作步骤(PROCEDURE)
整体介绍
打开CCP4i软件后,会出现如图 1所示界面。首先要设置project的名称及存储位置,本次举例的名称为“FXJ”。软件运行过程中需要的文件和输出的文件都会在该project中。如图 2所示,常用的有5个模块:数据处理,分子置换解析结构,结构模型的修正,结构模型的分析及map相关操作。
图 1 CCP4i软件打开后的界面
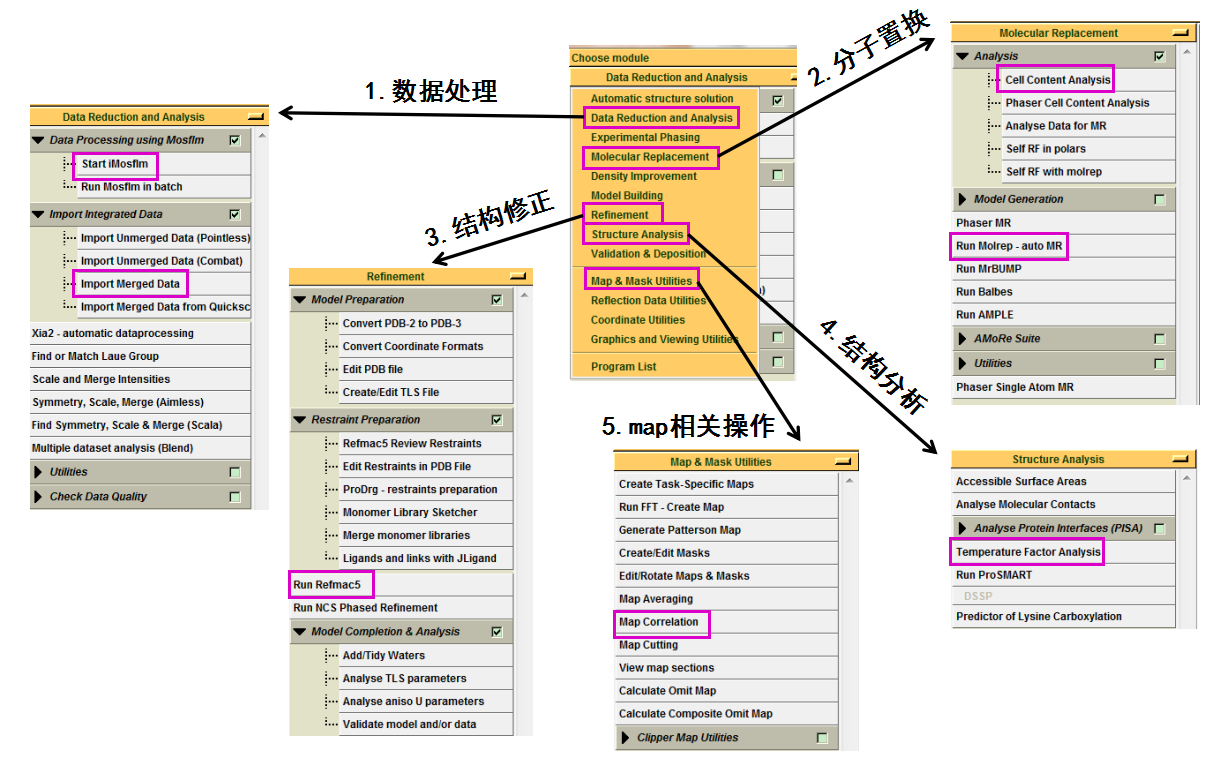
图 2 CCP4i软件中常用的5种模块
数据处理(一般不用iMosflm处理数据,简单了解一下)
1. 先在“Data Reduction and Analysis”下拉菜单中点击“Start iMosflm”,接着会出现如图 3所示的界面。
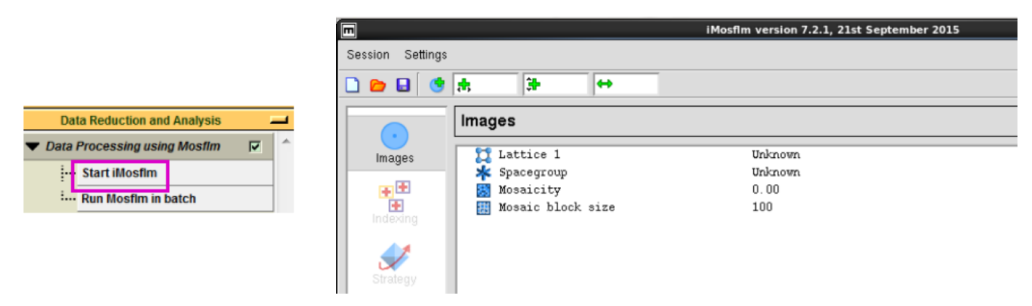
图 3 数据处理“Data Reduction and Analysis”界面
2. 点击图 4红框所示的图标,选择要处理的衍射画面文件。
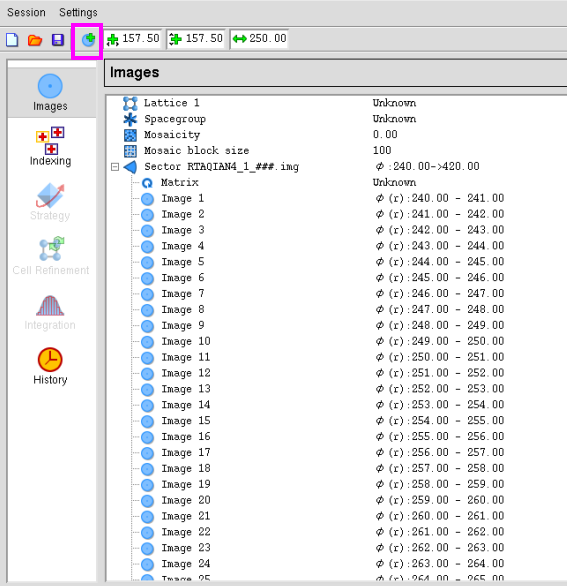
图 4 数据处理中衍射画面的输入
3. 点击图 5中的“Images”,可以查看衍射画面上衍射点的分辨率及衍射点的质量。
图 5 数据处理中衍射画面的查看
4. 点击“Indexing”,可以对衍射画面进行指标化(图 6图 7)。
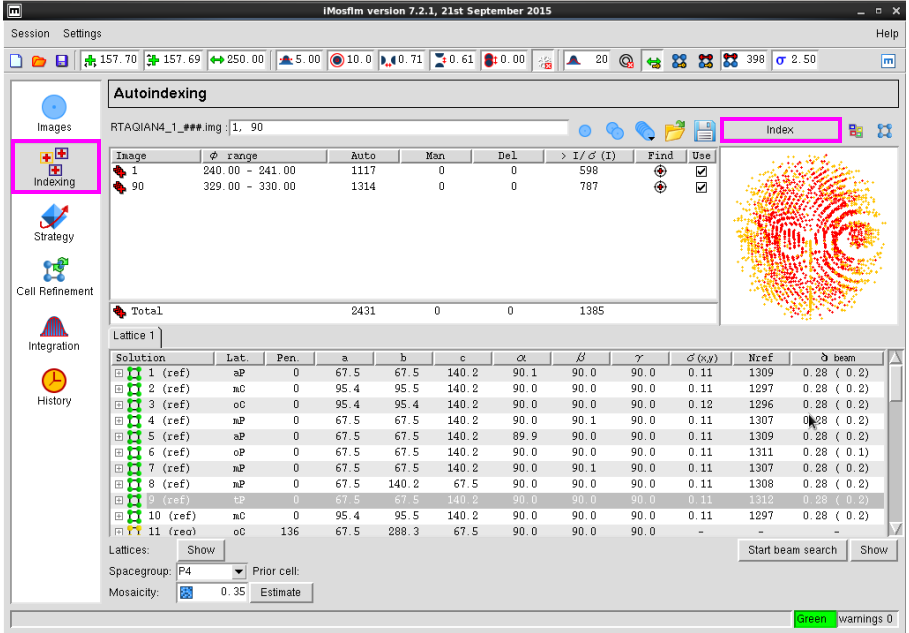
图 6 数据处理中Indexing界面
图 7 数据处理中Index过程
5. 在“Strategy”中可以查看参数(图 8)。

图 8 数据处理中“Strategy”界面
6. 在“Cell Refinement”中可以设置一些参数进行修正(图 9)。
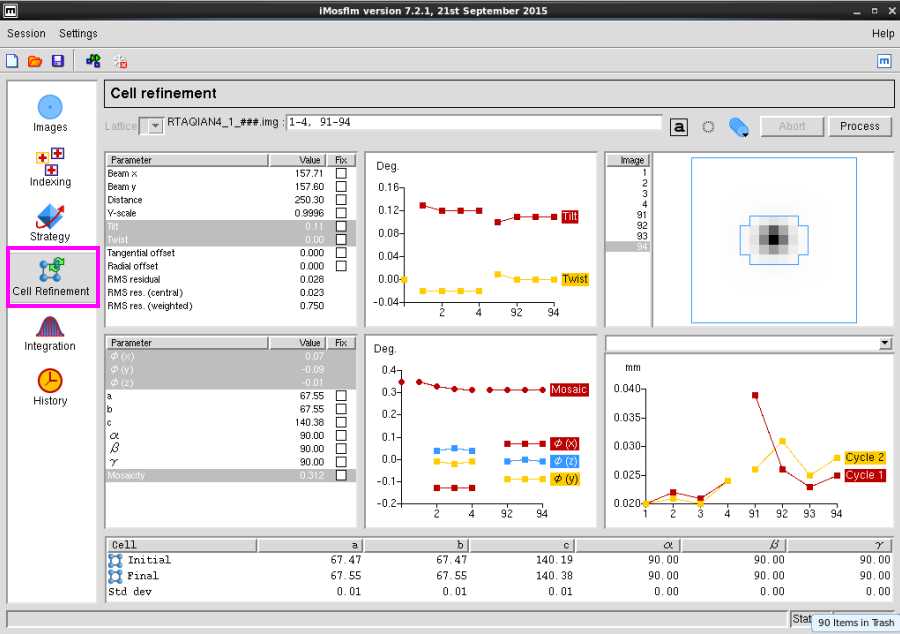
图 9 数据处理中Cell Refinement界面
7. 点击“Intergration”进行强度积分(图 10)。
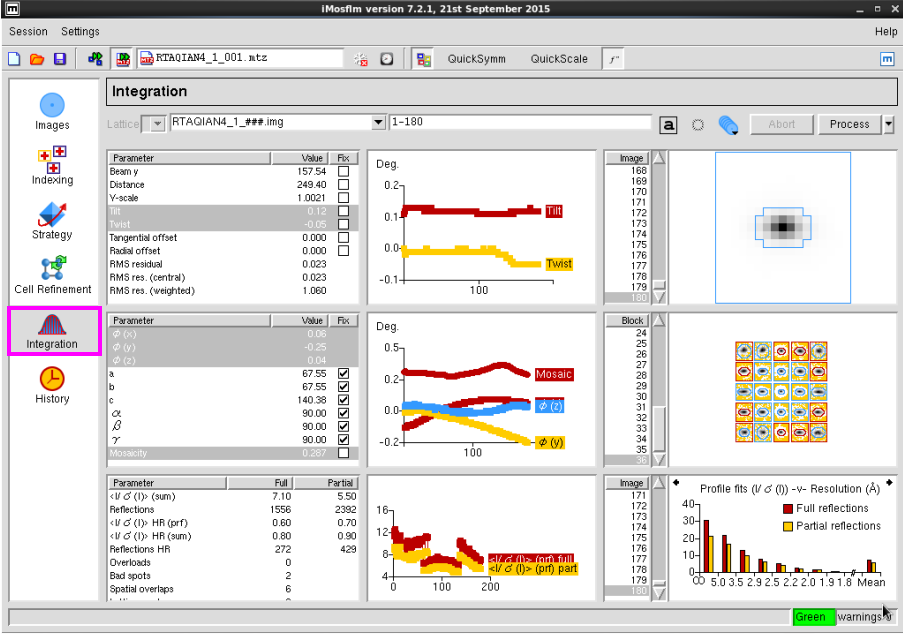
图 10 数据处理中Intergration界面
8. 在“History”中可以查看操作过程(图 11-图 12)。
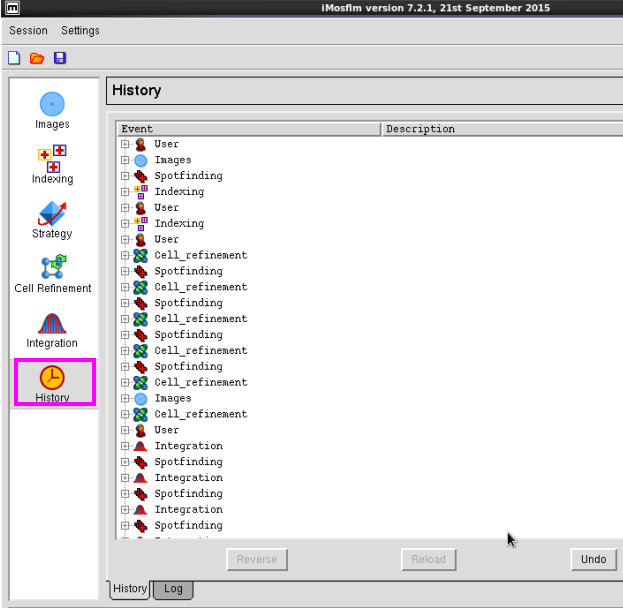
图 11 数据处理中History界面
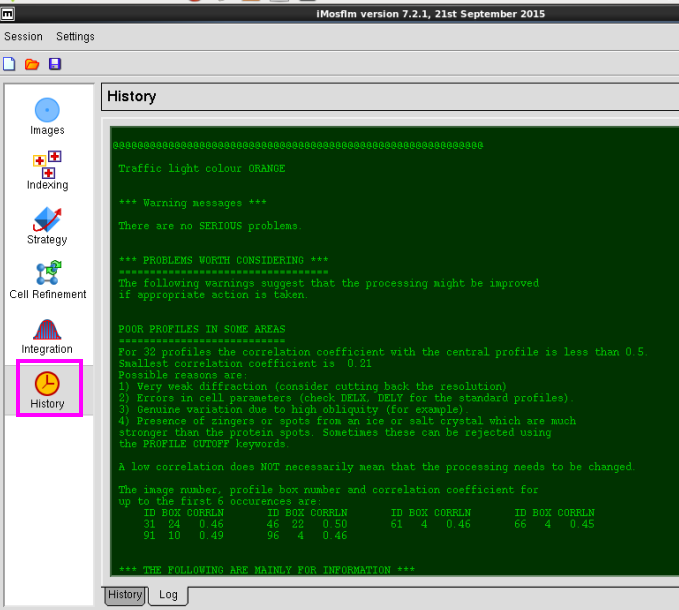
图 12 数据处理中History的log文件
9. 可以将使用iMosflm或HKL2000或XDS处理的sca文件,转化成mtz文件。具体操作为:在图 13所示的界面中,先点击“Import Merged Data”,可以输入“Job title”(本次举例未输名称),然后输入sca文件,软件会自动输出mtz文件的名称。(如果是反常散射数据,选中“Use anomalous data”)。一些参数会自动出现,如空间群,晶胞参数。然后点击“Run”,运行。
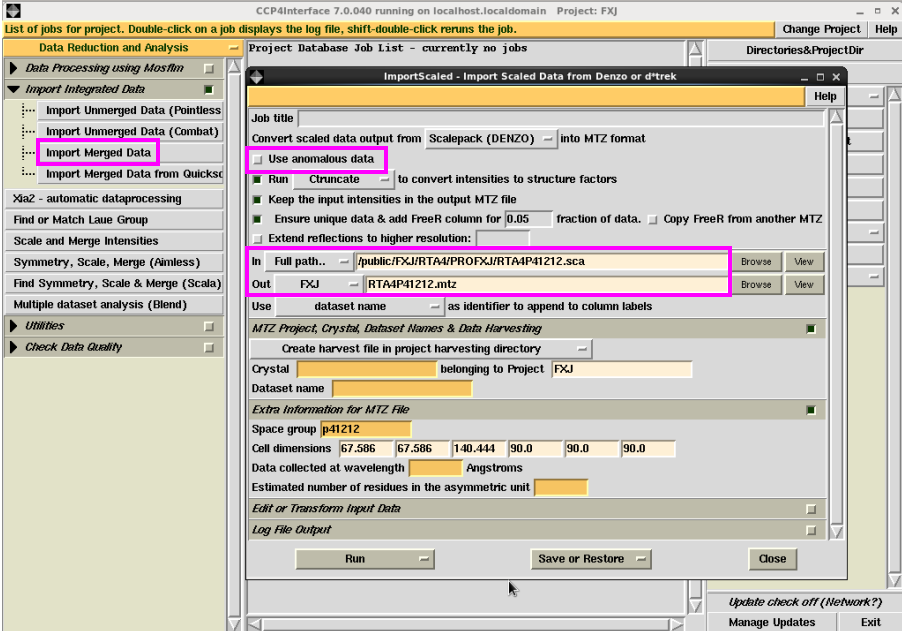
图 13 数据处理中Import Merged Data界面
10. 运行结束后,会显示本次job是“FXJ”project中运行的第一个job(图 14)。“FINISHED”说明运行成功并结束。在右边“View Files from job”可以查看log文件,里面有运行过程及最终的一些参数。如果本次运行结果出现“FAILED”,说明输入的一些参数或者文件本身有问题,可以根据log文件的说明查找原因改正。
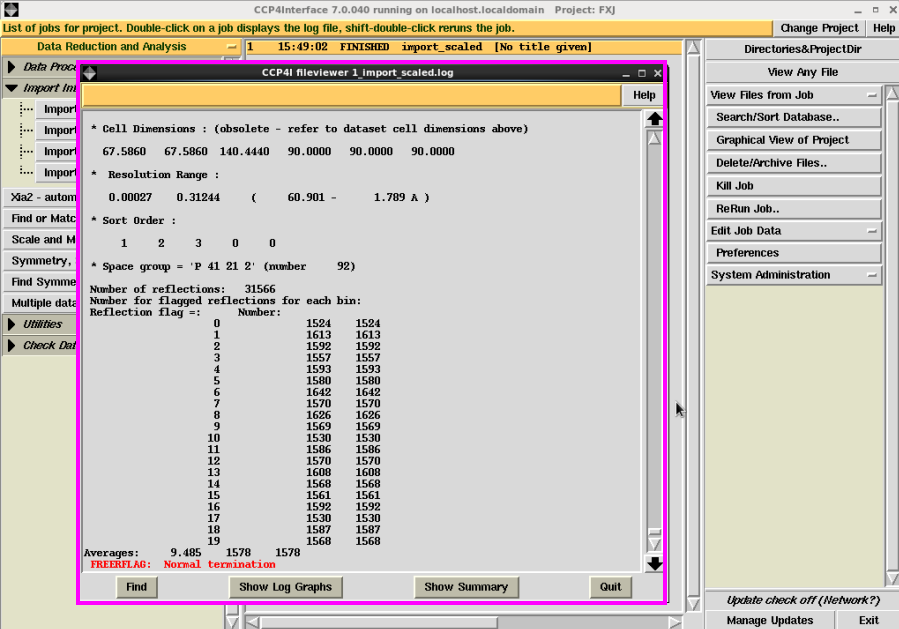
图 14 数据处理中Import Merged Data运行结束的log文件
分子置换(MR)
1. 首先在“Molecular Replacement”中点击“Cell content Analysis”,确定晶胞中一个不对称单位中的分子数(图 15)。输入mtz文件,选择使用蛋白残基数还是分子量,本次举例选择的是残基数,然后点击“Run Now” ,会显示出晶胞的体积及分子数。一般选择溶剂含量在50%左右对应的分子数,如本次例子中选择溶剂含量为53.96%,对应的分子数为1。可以作为后期解析的结构模型中的分子数的参考。
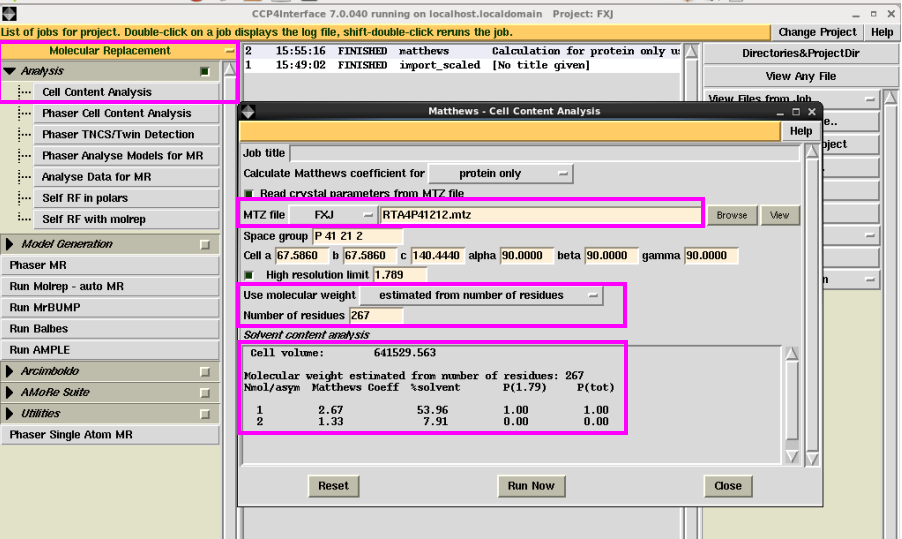
图 15 分子置换中Cell content Analysis界面
2. 点击“Run Molrep-auto MR”中,输入mtz和模型pdb文件后,点击“Run”,运行(图 16)。
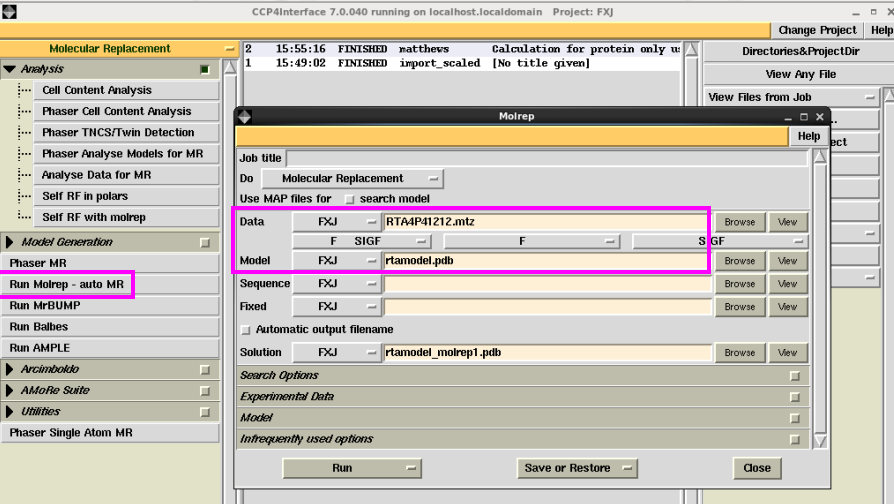
图 16 分子置换中Run Molrep-auto MR界面
3. 查看MR后的log文件,分析是否有解(图 17)。一般Contrast值大于3说明有解。
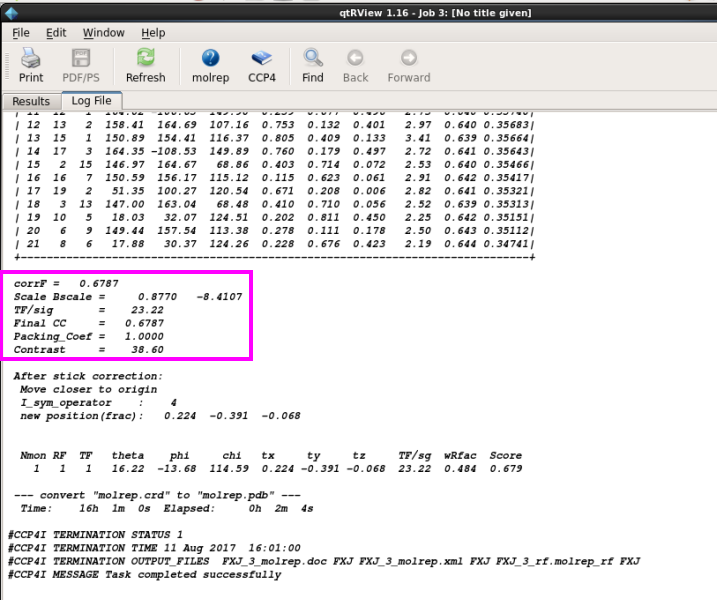
图 17 分子置换中Run Molrep-auto MR后的log文件
4. 在“Results”中,也可以用CCP4MG和Coot查看置换的pdb文件(图 18图 19)。
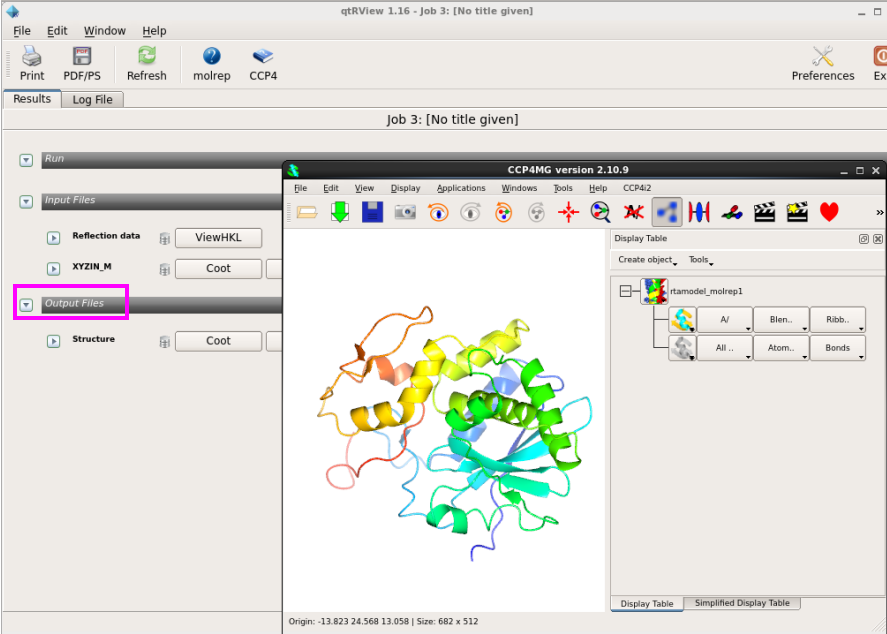
图 18 分子置换中CCP4MG查看结构模型
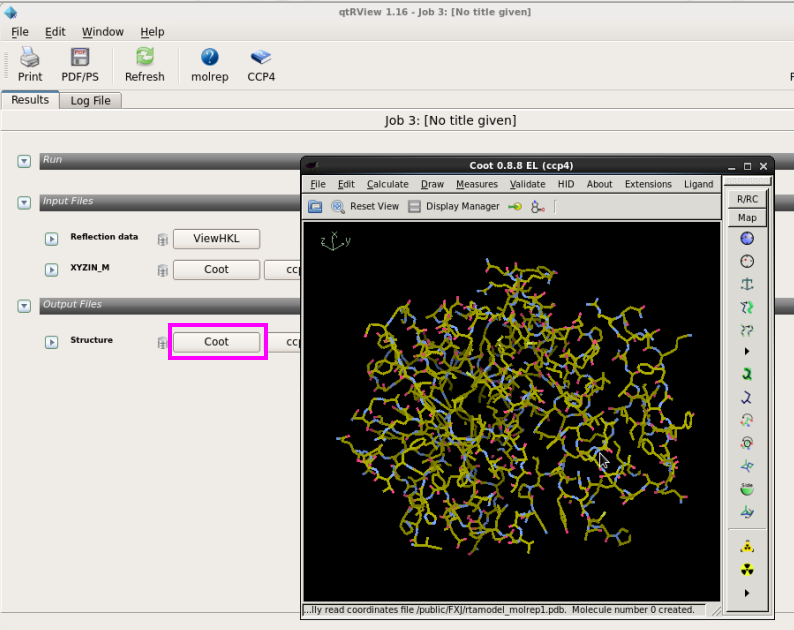
图 19 分子置换中Coot查看结构模型
结构模型的修正
1. 在“Refinement”中点击“Run Rfmac5”进行结构模型的修正,一般先进行刚体修正。具体操作为:在图 20中“Do”中选择“rigid body refinement”,然后输入mtz文件和分子置换后的pdb文件。输出文件软件会自动给出。在该界面中还可以修改分辨率范围,根据不同的需要可以修改其他参数,一般可以选择默认。然后点击“Run”运行即可。查看刚体修正的Rfactor和Rfree。一般只进行刚体修正的R值降低幅度较小(图 21)。此处也可以用Coot查看修正后的模型和电子密度吻合情况,也可直接用打开的Coot进行手动修正(图 22)。
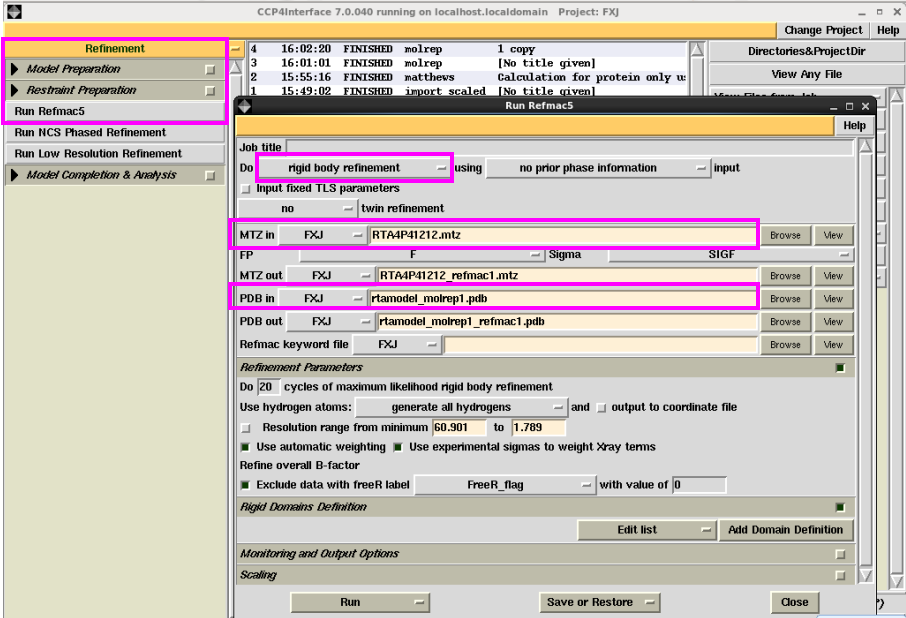
图 20 结构模型修正中刚体修正界面
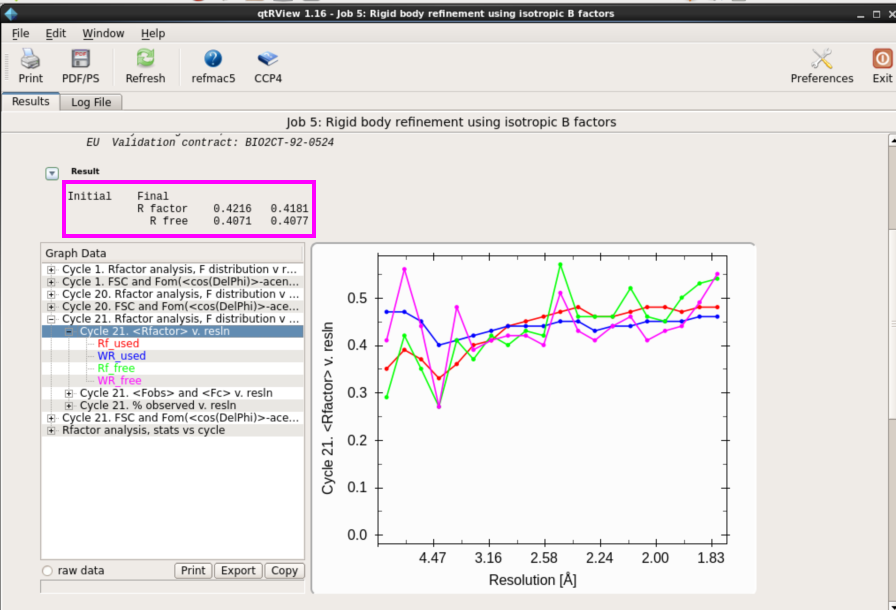
图 21 结构模型修正中刚体修正后的结果
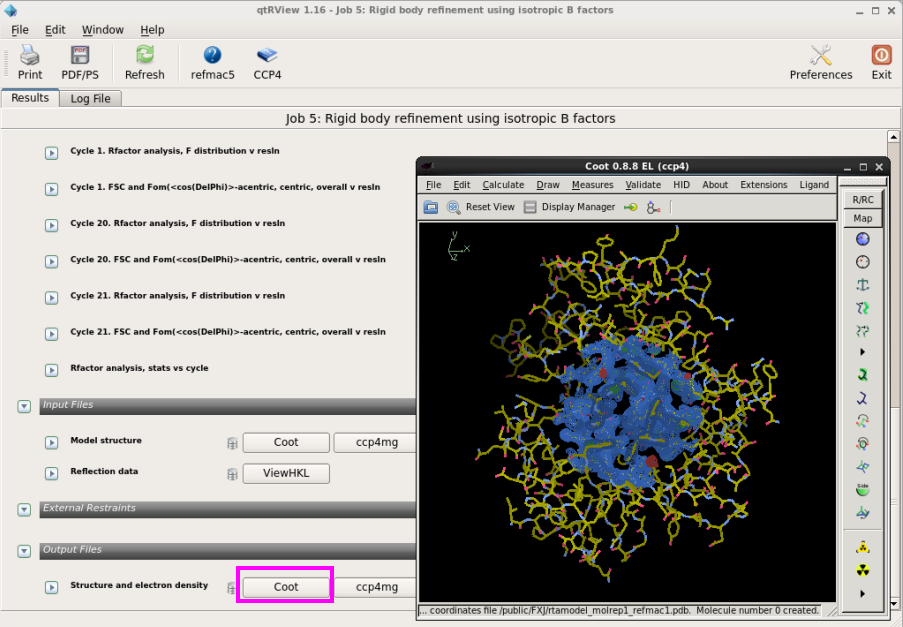
图 22 结构模型修正中Coot查看模型和电子密度吻和情况
2. 刚体修正结束后,再进行温度因子修正(图 23)。此时在“Do”处选择“restrained refinement”,输入的文件是上一轮刚体修正的pdb文件,mtz文件还是最原始的文件。在“refine”处选择“isotropic”,点击“Run”。查看修正的Rfactor和Rfree,是否在降低。一般温度因子修正后的R值降低幅度很大。同时查看“Rms bondlength”和“Rms BondAngle”值是否在允许的误差范围内(图 24)。
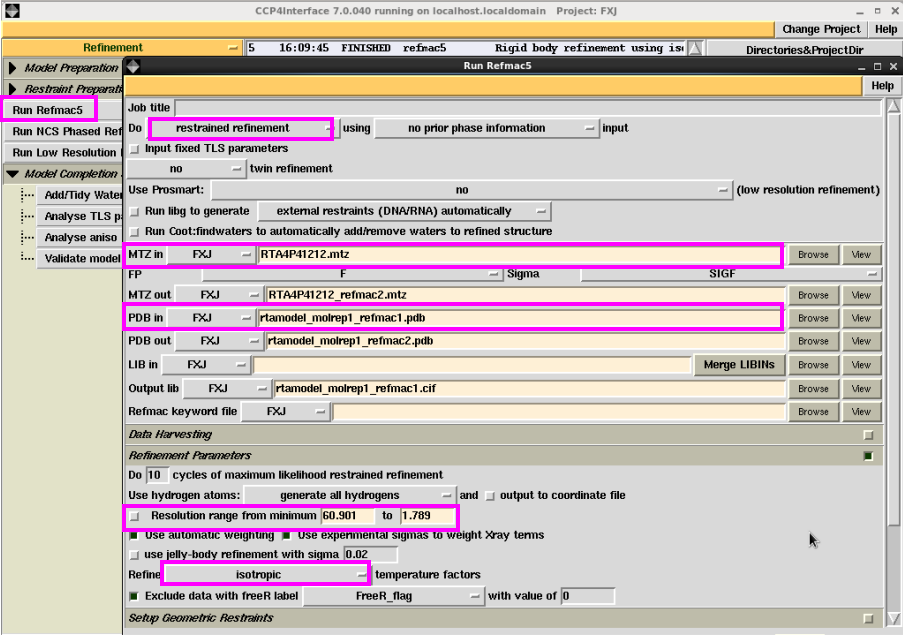
图 23 结构模型修正中的温度因子修正界面
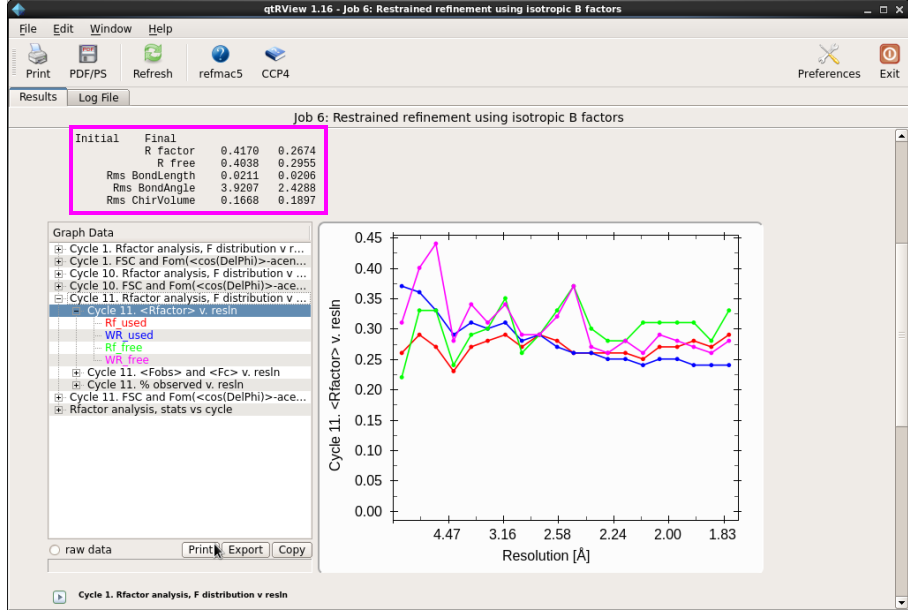
图 24 结构模型修正中温度因子修正结果
3. 也可以先进行坐标修正(overall)再进行温度因子(isotropic)修正(图 25图 26)。接着进行温度因子修正。R值略低于直接用温度因子修正的结果,但差别不大(图 27-图 28)。第3步和第2步属于2种不同的操作,结果类似。
图 25 结构模型的overall修正界面

图 26 结构模型的overall修正结果
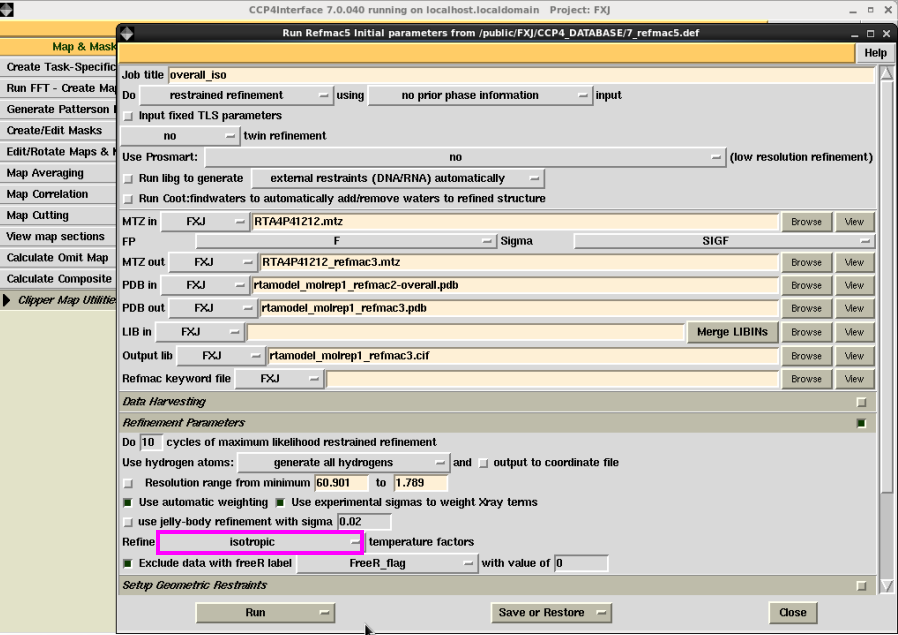
图 27 结构模型修正中温度因子修正界面
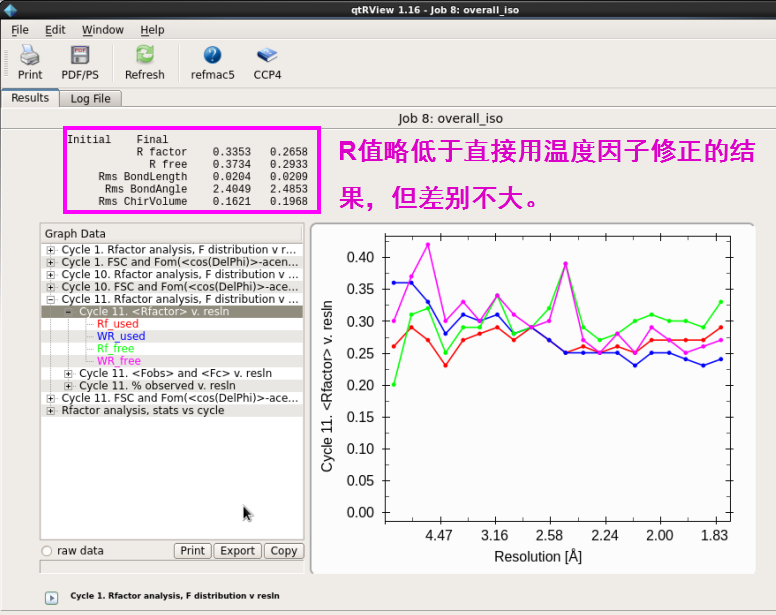
图 28 结构模型修正中温度因子修正结果
结构模型的分析
1. 温度因子分析。在“Structure Analysis”下拉菜单中“Analyse Protein interfaces(PISA)”中点击“Temperature Factor Analysis”,输入修正的pdb文件,然后点击“Run”。运行结束后可以查看每条链上氨基酸残基主侧链对应的温度因子,以及所有原子的数目和总的温度因子(图 29图 30)。
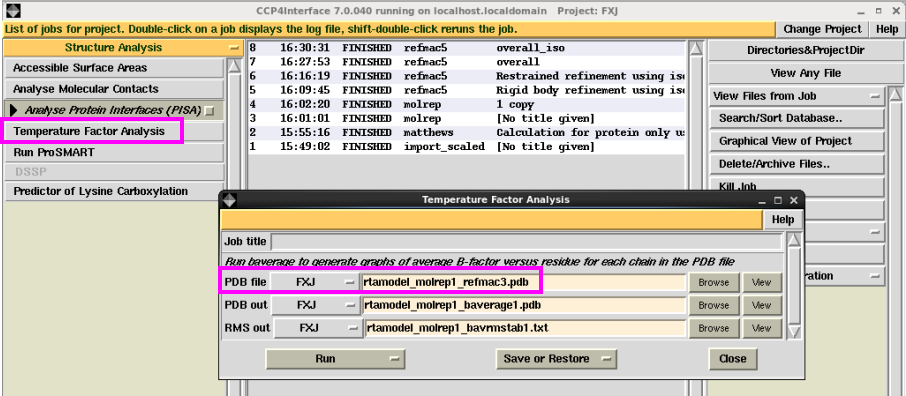
图 29 结构模型的温度因子分析界面
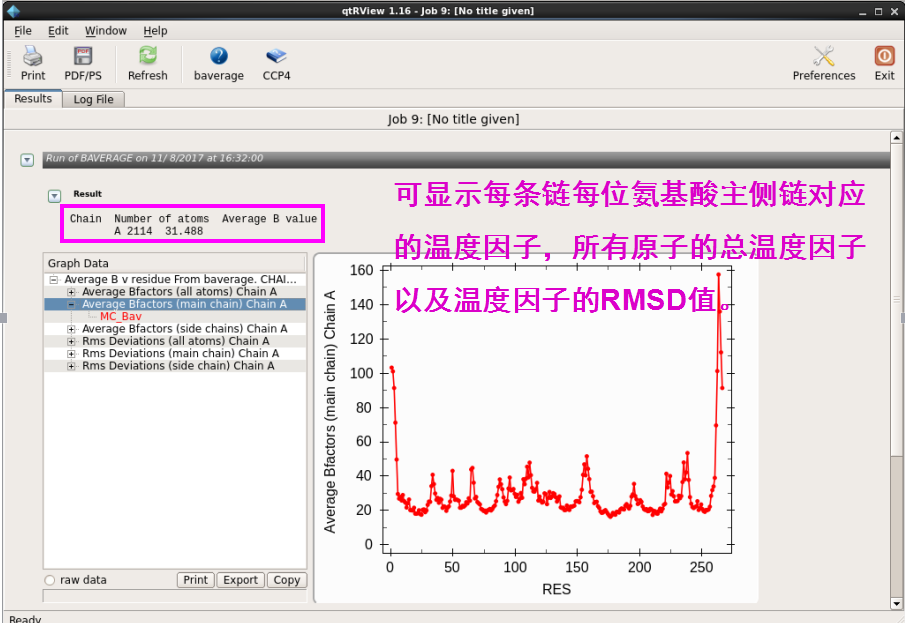
图 30 结构模型的温度因子分析结果
2. map相关操作。主要介绍时空间相关系数分析。在“Map & Mask Utllities”下拉菜单中选择“Map Correlation”,输入mtz文件和pdb文件,点击“Run”。运行结束后,可以查看每条链上的氨基酸残基对应的实空间相关系数,一般大于0.8是比较好的(图 31图 32)。
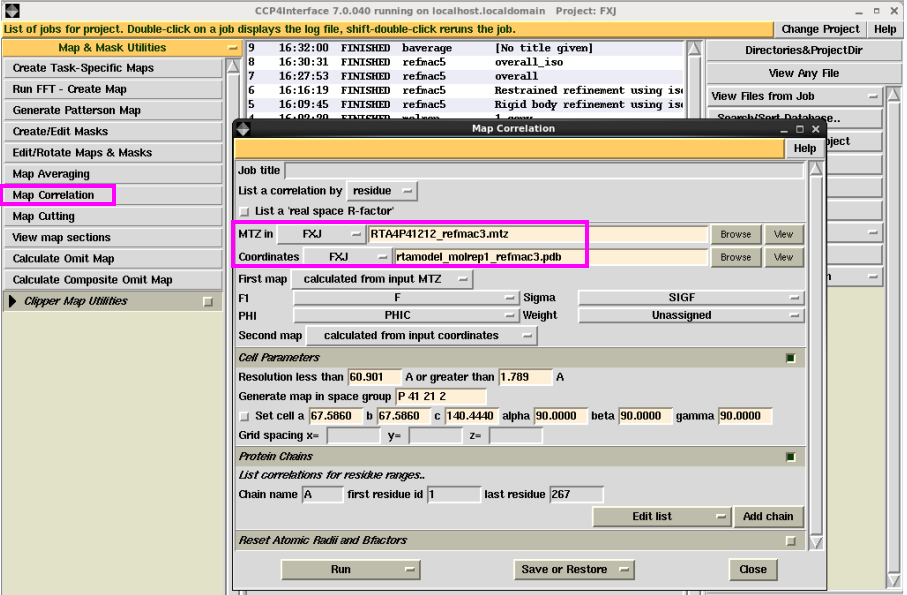
图 31 结构模型的实空间相关系数分析界面

图 32 结构模型的实空间相关系数分析结果
5.13.3 Phenix基本操作
简介(INTRODUCTION)
PHENIX(Python-based Hierarchical ENvironment for Intergrated Xtallography)是一种自动解析生物大分子晶体结构的综合软件。
操作步骤(PROCEDURE)
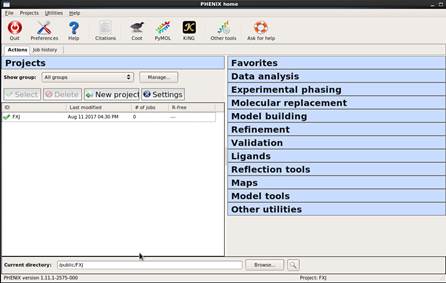
整体介绍
打开Phenix后首先出现所示界面,点击“OK”后,设置project的名称(如 “FXJ”)以及存储位置(如“/public”),保存后进入图 34所示界面。界面右边显示一些常用的操作。图 35显示的是常用的操作,单击某操作后会有下拉菜单选项。
图 33 打开Phenix后的初始界面
![]()
![]()
- 图 34 Phenix设置project后的界面
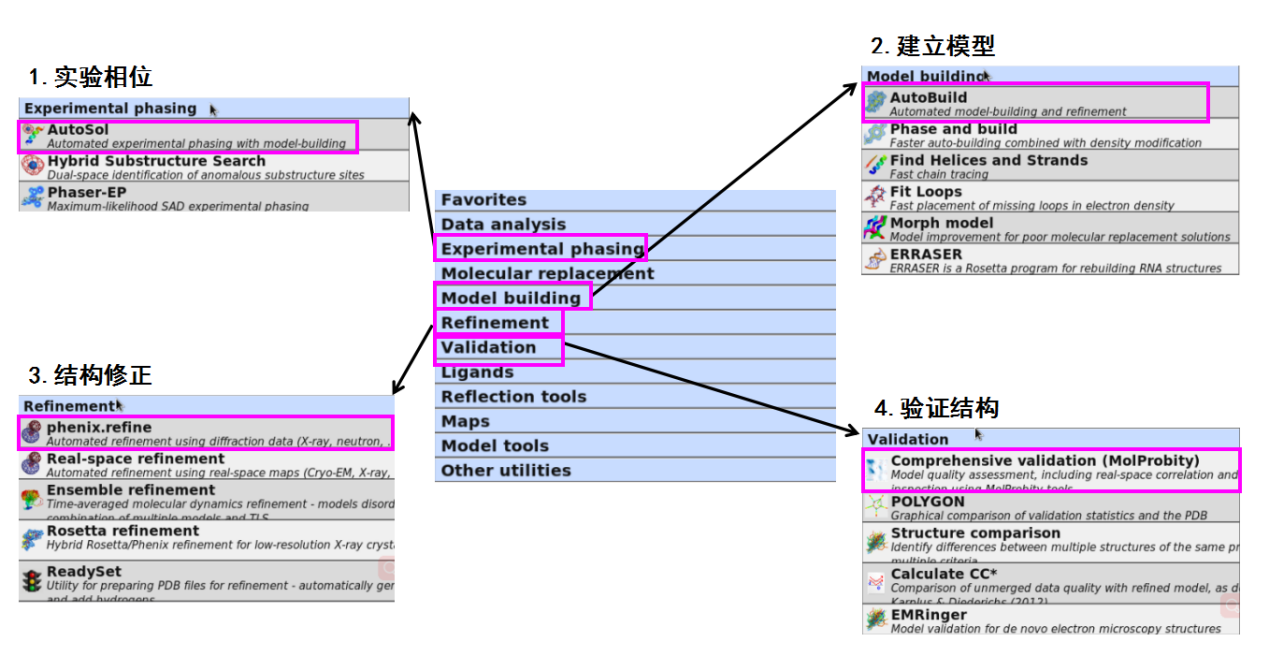
图 35 Phenix中常用操作
实验相位(以硒代数据为例,获得硒代相位)
1. 单击“AutoSol”后,输入sca和seq文件,波长,Se及数目(图 36)。其他参数可选填(图37)
2. 点击“Run”,开始运行。每次的运行时间不一致,本次运行约1 h 40 min(图 38)。

图 36AutoSol的Input files界面
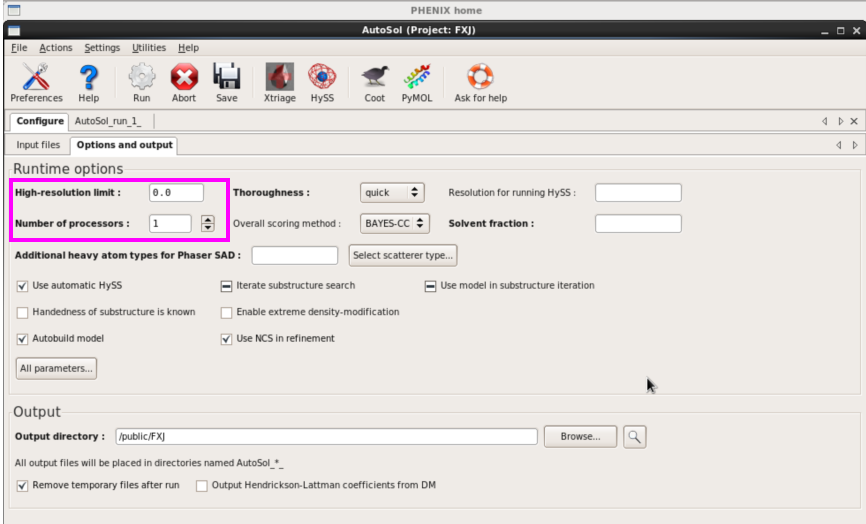
图 37 AutoSol的options and output界面
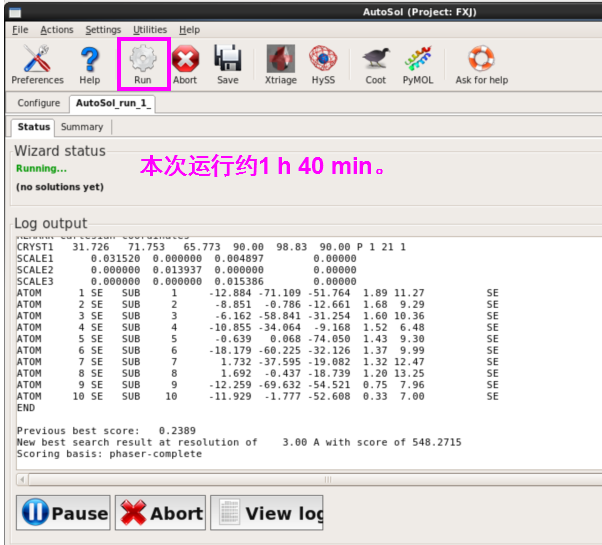
图 38 AutoSol的运行界面
3. 查看结果。当显示“FINISHED”时,说明运行结束(图 39)。在“Status”栏中可以查看se原子的数目(Sites),空间群(P1211),FOM值,氨基酸残基数(Residues),几条链(Chains),CC值(Model CC),Rwork和Rfree。

图 39 AutoSol运行结束后的States界面
4. 在“Summary”中也可以查看相关参数,同时可以看到添加的水分子数目(图 40)。
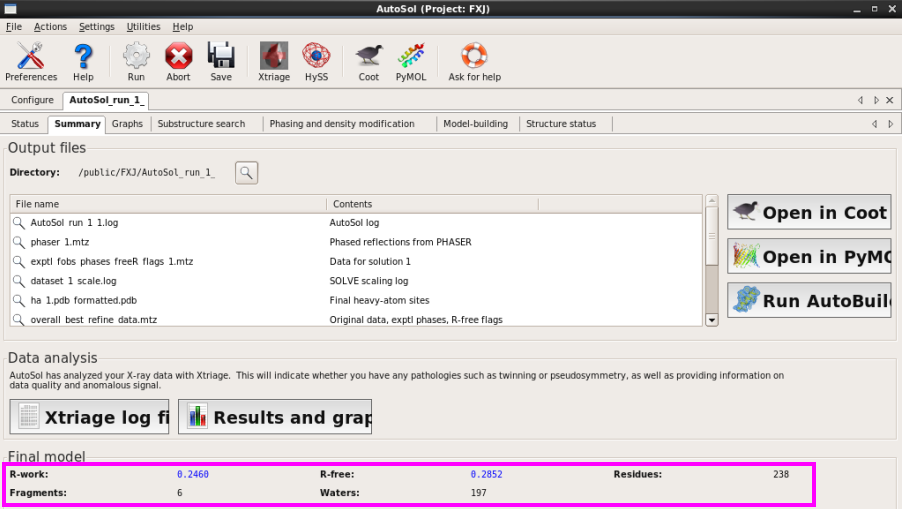
图 40 AutoSol运行结束后的Summary界面
5. 单击“Open in coot”,可以查看产生的结构模型和电子密度(图 41)。也可以从此处开始手动修正结构模型。
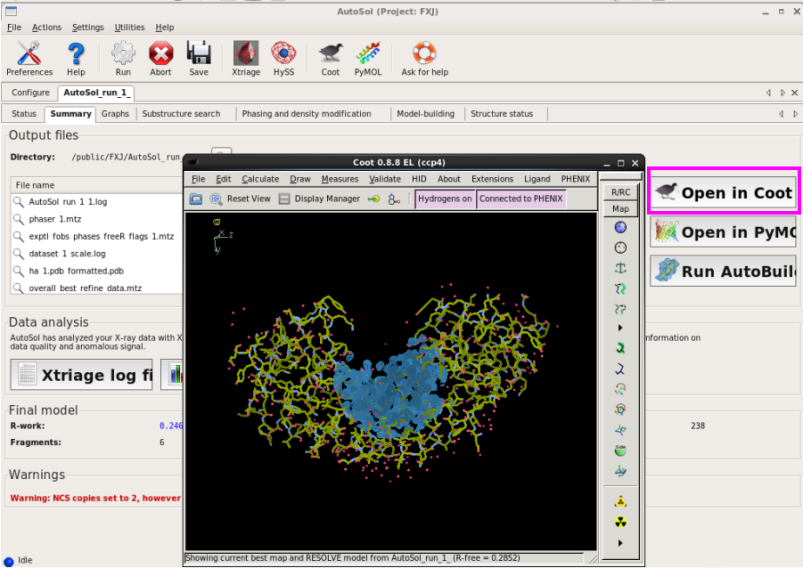
图 41 AutoSol运行结束后用Coot查看界面
6. 单击“Open in PyMOL”,可以查看产生的结构模型(图 42)。也可以进行简单的结构分析。
图 42 AutoSol运行结束后用PyMol查看界面
7. 在“Graphs”栏,可以查看产生的各种图形参数(图 43)。
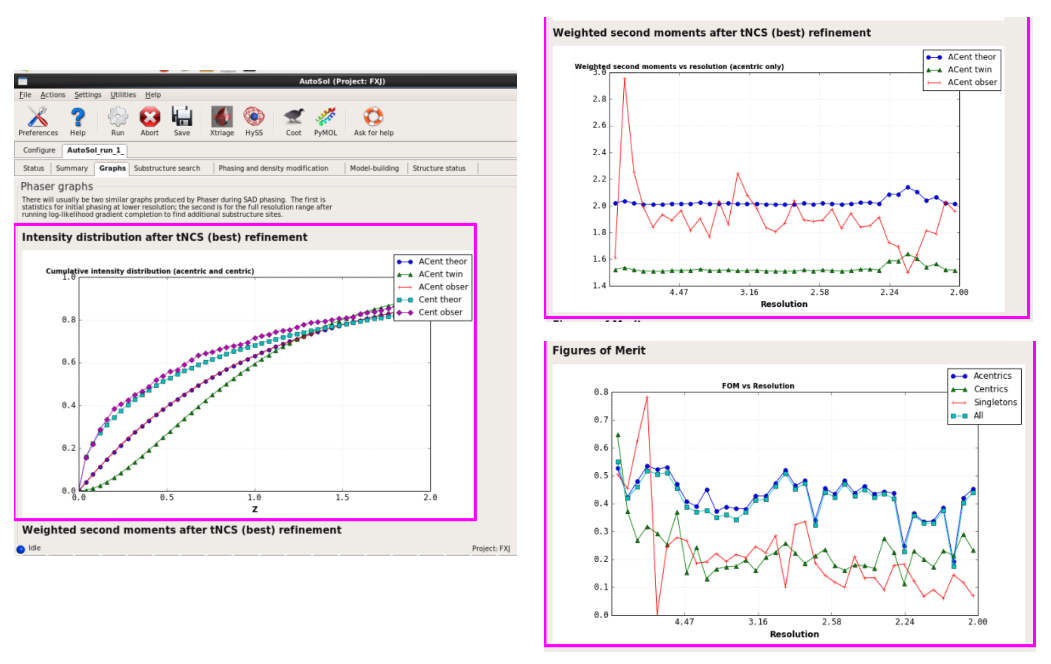
图 43 AutoSol运行结束后的Graphs界面
8. 在“Substructure search”栏可以查看substructure的解及相关参数。如每个Se原子的占有率(图 44)。
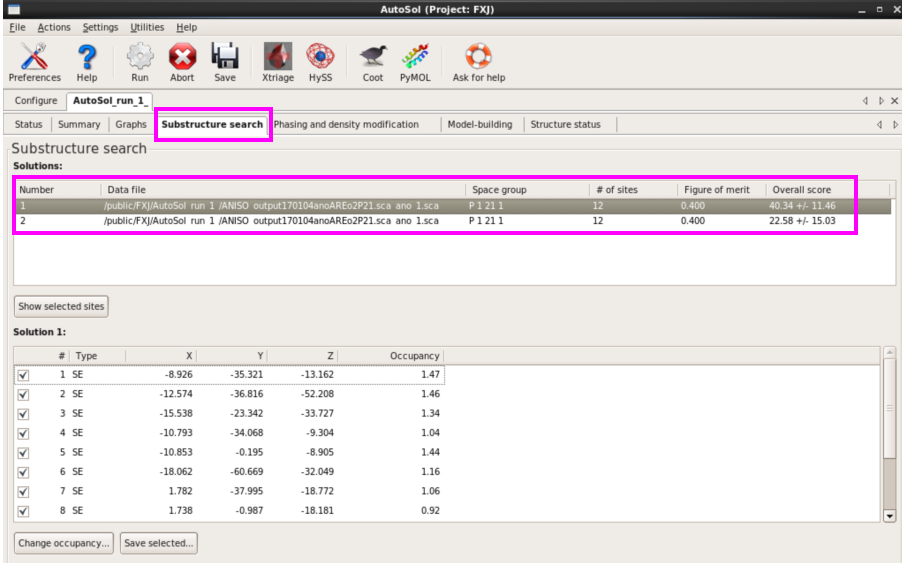
图 44 AutoSol运行结束后的Substructure search界面
9. 在“Phasing and density modification”栏,可以查看每个解的R-factor,Corr. of local RMS density(图 45)。还可以查看每个Se原子在电子密度图中的位置()。
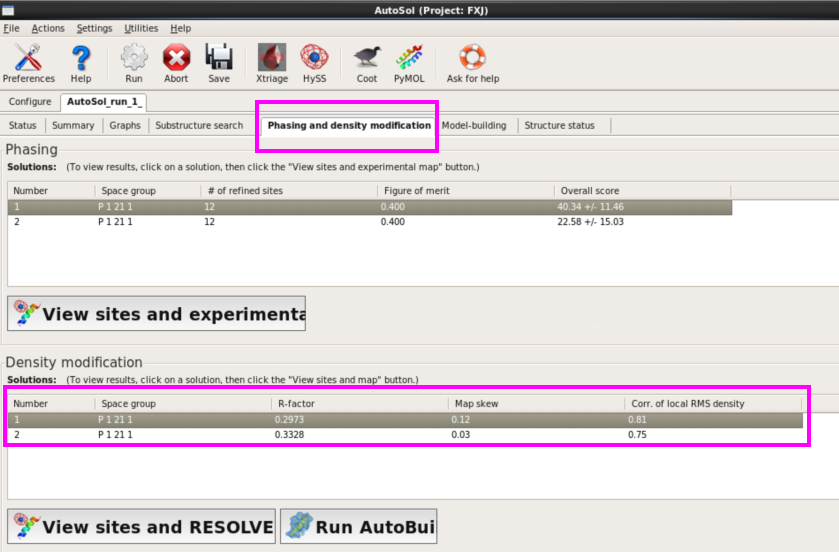
图 45 AutoSol运行结束后的Phasing and density modification界面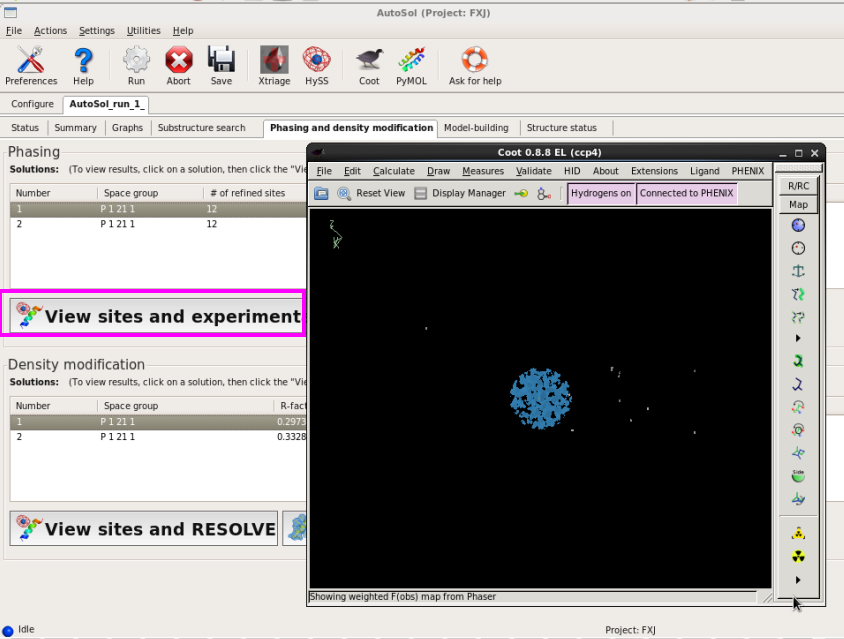
图 46 AutoSol运行结束后显示的硒原子相位
10. 在“Model-building”栏,可以看到软件给出的建立模型的解,本次运行只有一个模型(图 47)。模型的氨基酸残基数,R值都显示出来了。
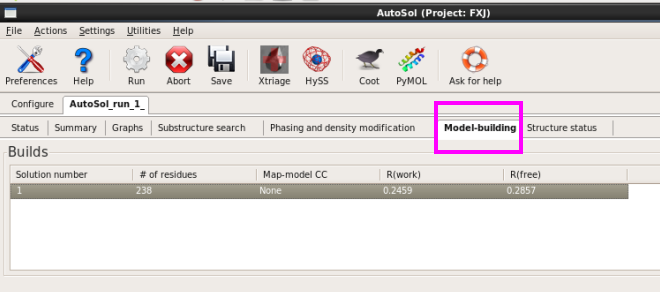
图 47 AutoSol运行结束后的Model-build界面
11. 在“Structure status”栏,可以查看模型已建立的氨基酸残基(图 48)。
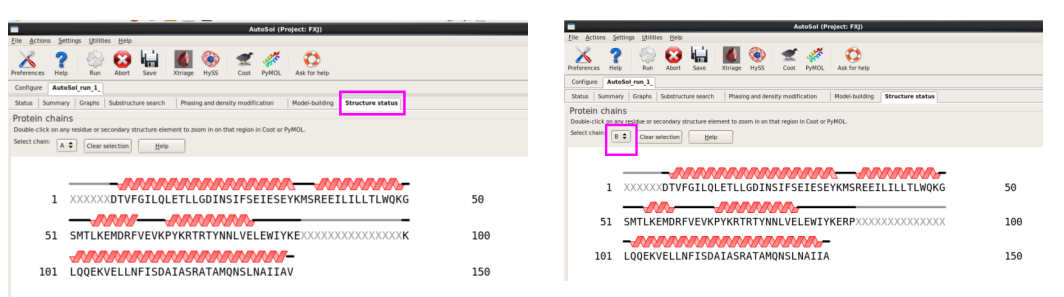
图 48 AutoSol运行结束后的Structure states界面
建立模型
1. 第一种方法:在本界面进行(图 49)。输入seq,mtz以及pdb文件,可以输入job title以及输出存储位置(图 50)。运行“Run” 。查看运行结果。本次运行约8 h。查看结果同模型建立,可以打开每一栏进行查看(图 51图 53)。

图 49 Phenix中Model building界面
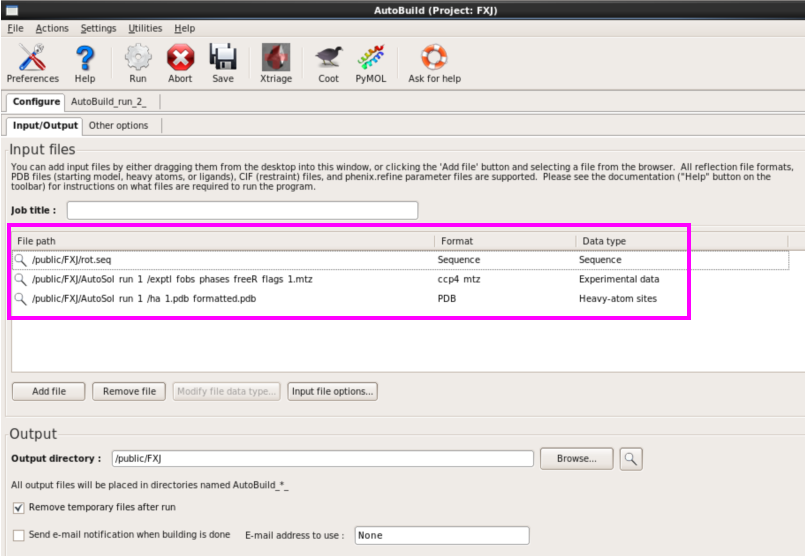
图 50 Model building中AutoBuild界面
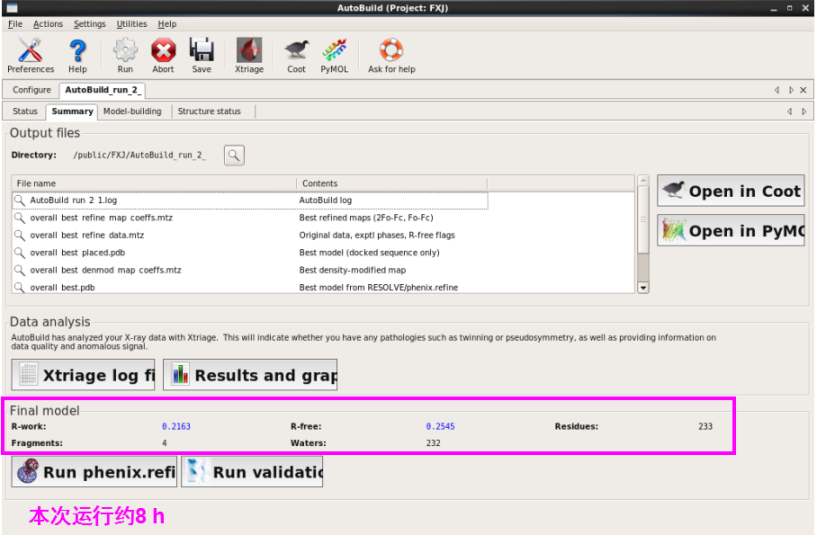
图 51 AutoBuild运行结束界面
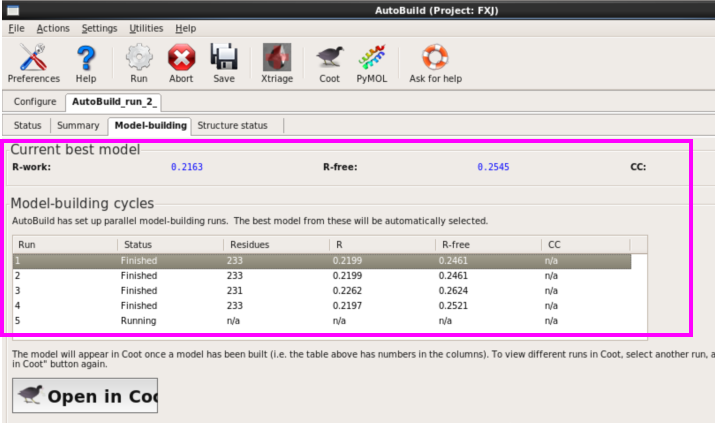
图 52 AutoBuild运行结束后的Model-building界面 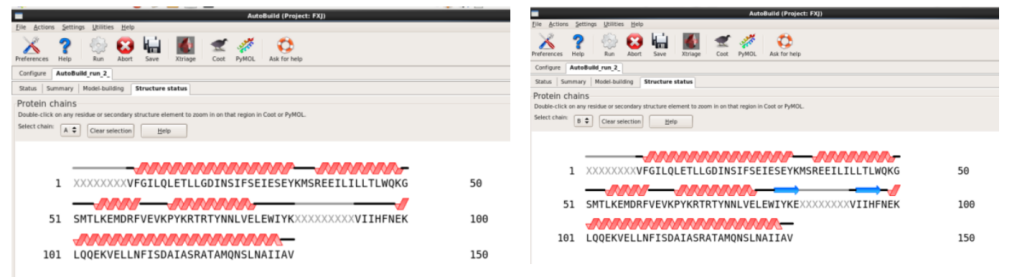
图 53 AutoBuild运行结束后的Structure states界面
2. 第二种方法:在AutoSol结果中直接Run AutoBuild(图 54)。结果类似(图 55)。
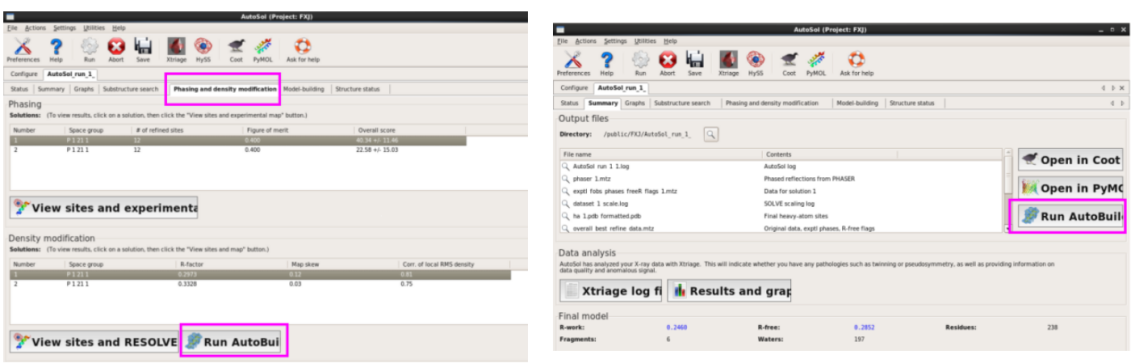
图 54 AutoSol中的Run AutoBuild
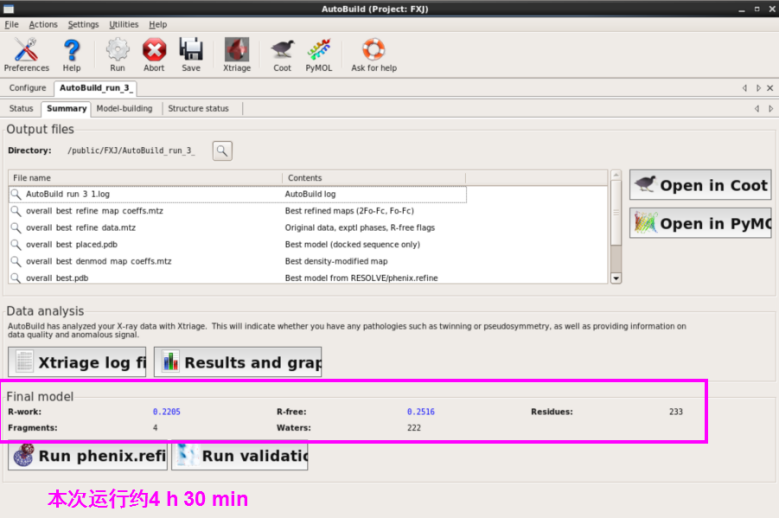
图 55 AutoSol中的Run AutoBuild结束界面
结构修正
1. 输入mtz文件和pdb文件。如图 56所示,设置修正需要的参数,一般先选择默认(图 57)。
图 56 Refinement的Input界面
图 57 Refinement的Refinement settings界面
2. 然后点击运行“Run”。可以看到运行过程及一些参数的变化(图 58)。

图 58 Refine的运行过程
3. 显示“Finished”说明运行结束(图 59)。
图 59 Refine的运行结束界面
4. 查看结果(图 60)。
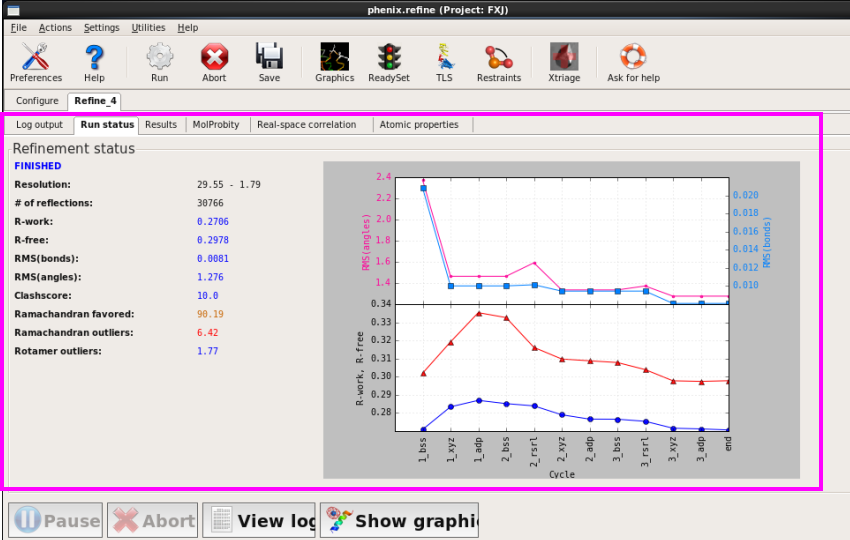
图 60 Refine的Run states界面
5. 在“Results”中可以看到产生相关mtz和pdb文件,R值以及键长键角RMSD值(图 61)。

图 61 Refine的Results界面
查看序列及二级结构组成,R值和CC值随分辨率壳层分布(图 62图 64)。
图 62 Refine结束后结构模型的序列查看界面
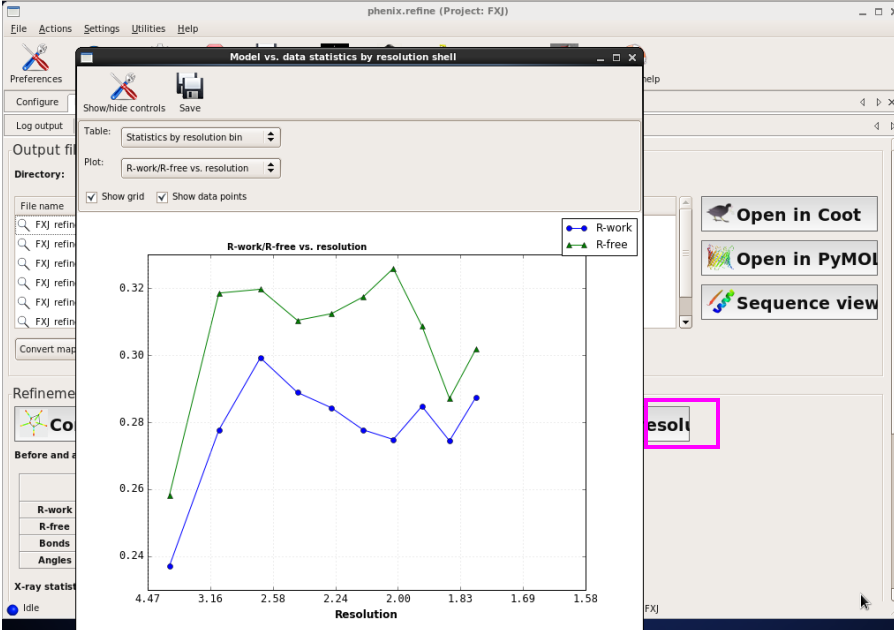
图 63 Refine结束后结构模型的R因子随分辨率壳层的分布情况
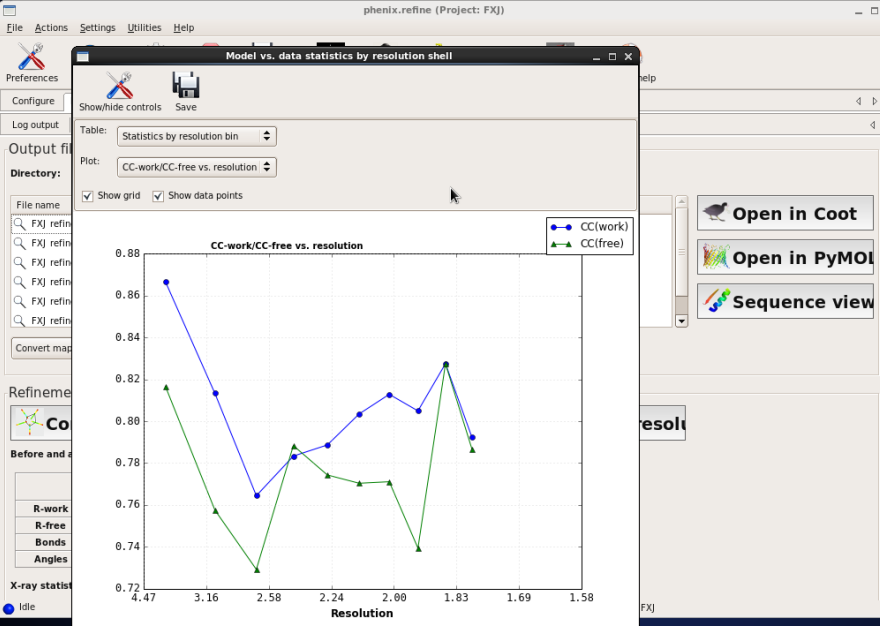
图 64 Refine结束后结构模型的CC值随分辨率壳层分布
7. 在“MolProbity”中查看Ramachandran图,几何结构参数以及过近接触的原子(图 65图 68)。
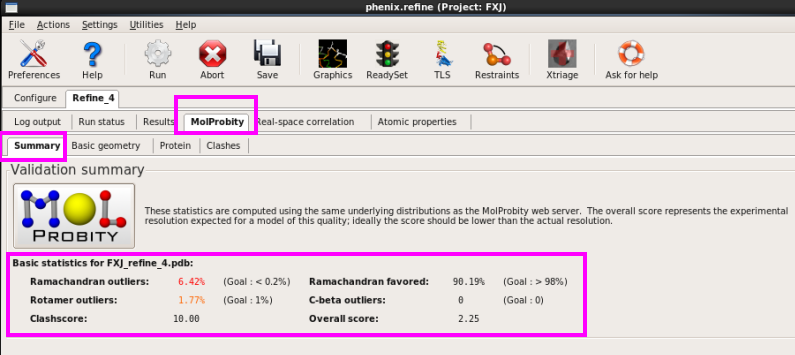
图 65 Refine结束后结构模型的Ramachandran图结果

图 66 几何结构参数
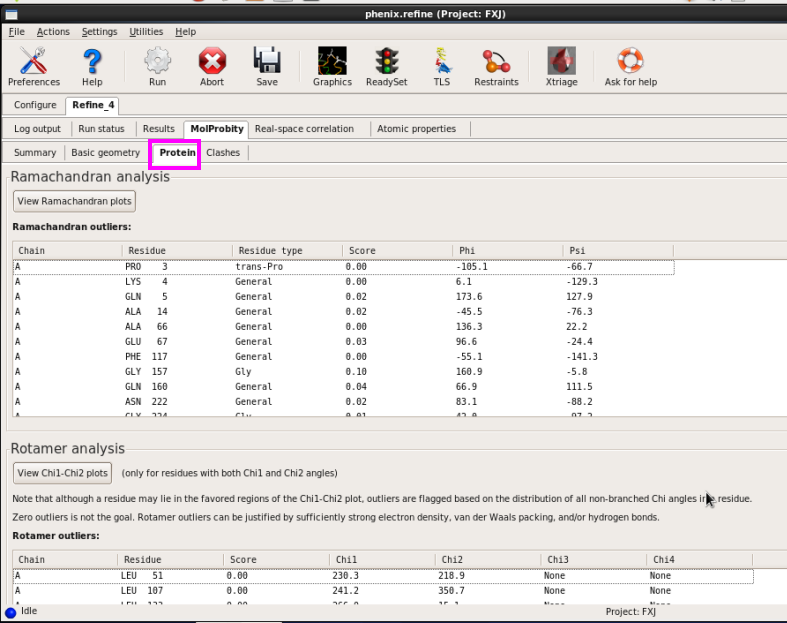
图 67 的Protein界面

图 68 Clashes界面
8. 在“Real-space correlation”中查看时空间相关系数(CC)(图 69图 70)。
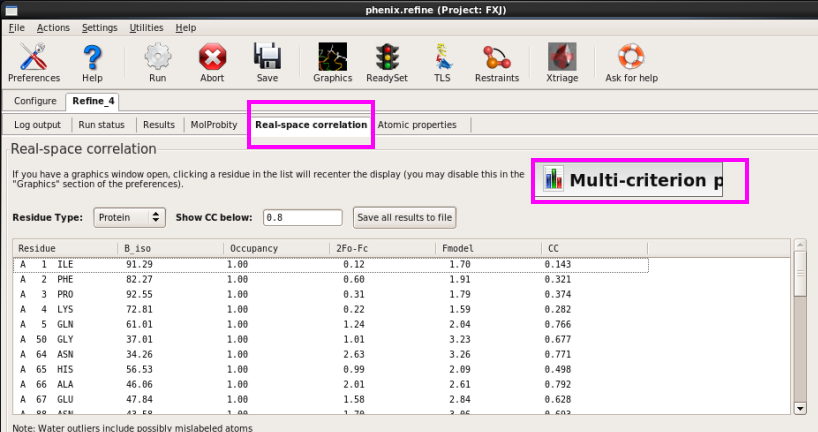
图 69 Real-space correlation界面

图 70 Real-space correlation中的Muti-criterion plot
9. 在“Atomic properties”中查看温度因子(图 71)。
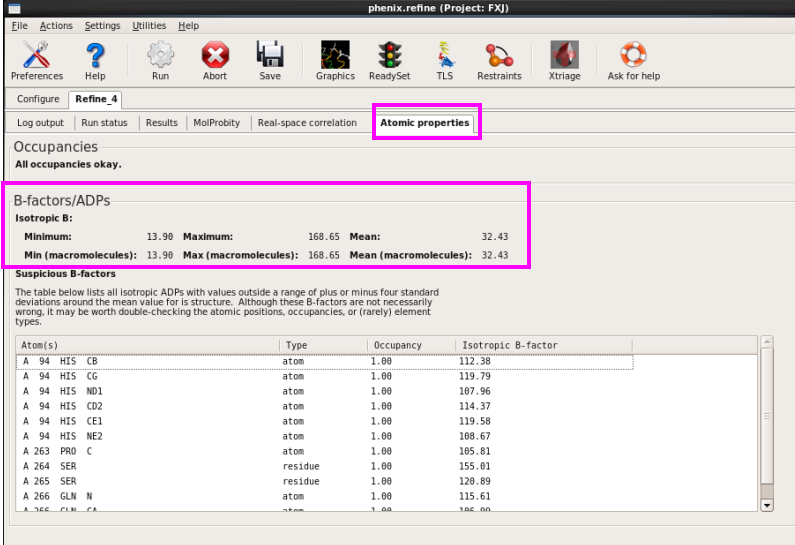
图 71 Atomic properties界面
验证结构
打开Validation界面,输入pdb和mtz文件并运行,运行结束查看结果(图 72图 75)。类似于结构分析中的各种参数。

图 72 Validation界面
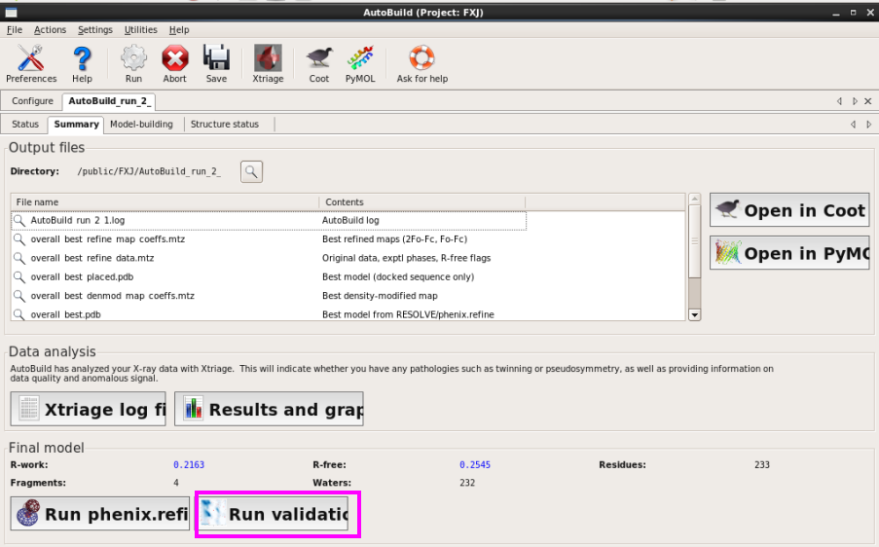
图 73 AutoBuild中的validation界面
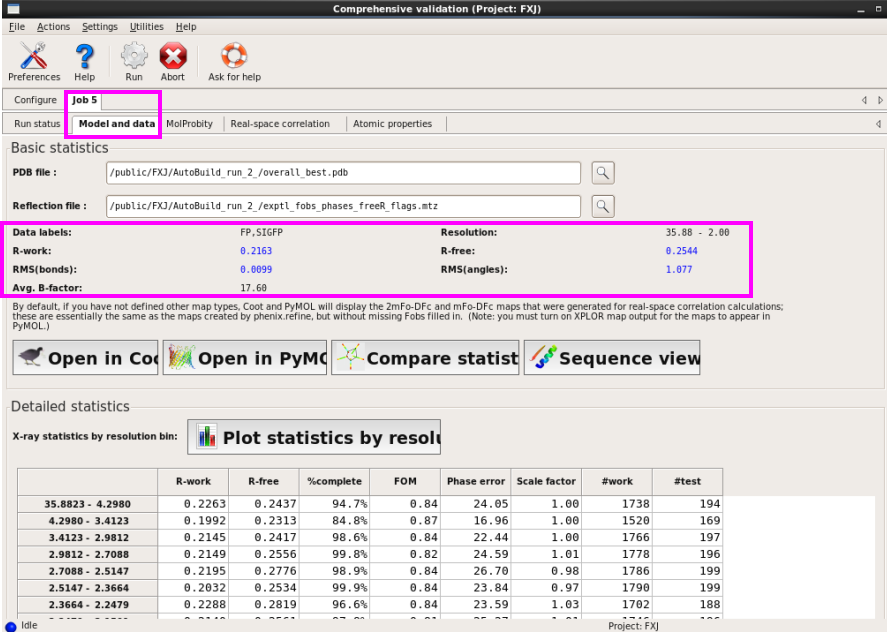
图 74 validation结束后的Model and data界面

图 75 validation结束后的MolProbity界面
5.13.4 Phenix.refine 常见问题和解答
简介(INTRODUCTION)
Phenix 是一个非常好的用于晶体学的软件包,由劳伦斯伯克利国家实验室Paul Adam联合剑桥大学,杜克大学和罗斯按拉莫斯国家实验室等团队,共同完成。其中最优秀的一款应用,就是phenix.refine它继承了Paul Adam当年主要参与开发的CNS软件包的很多优秀基因。Phenix.refine的主要作者都在Paul团队,包括Pavel Afonine 和Nicholas Sauter等。Phenix.refine是目前用于晶体结构自动修正最好的软件,特点是功能强大,包含simulated annealing等功能。并且年轻的作者团队,不断推出新版本和补丁。同时积极给全世界用户提供技术支持。下面是来自Phenix.refinment 开发团队的一些常见问题和解答。
目录
1. 我怎样才能使phenix.refine运行得更快?
目前有两种选择:
OpenMP并行化效率不是特别高,因为FFT需时少于典型运行时间的一半;提速通常为40%。当启用约束权重优化时,使用“nproc”进行process-level并行化非常有用,因为这些步骤可以作为多个单独的流程运行。在这些情况下,可以加速4~5倍。但是,使用默认参数的运行不会从设置'nproc'中获益。
这些选项有几个局限:
如上所述,标准并行化主要影响权重优化。如果不执行此操作,则大多数程序将以串行方式运行。
3. 我应该使用什么类型的实验数据进行细化?
可以在细化中使用振幅(F)或强度(I)(以任何文件格式),优先使用强度。支持反常和非反常数据;使用默认策略中的一个或另一个对模型质量似乎没有任何特别的好处。然而,反常数据可用于优化重原子的反常散射因子,并且将在输出的MTZ文件中自动创建反常差异map系数。由于这些原因,建议使用反常数据,但一般来说R因子是相似的。
4. 我应该使用phenix.refine输出的MTZ文件作为下一轮细化的输入吗?
当对不包含R-free flag的数据集进行优化时才需要这么做,让phenix.refine生成一个新的测试集。在这种情况下,您应该使用以“_data.mtz”结尾的文件进行以后的所有优化。不需要在每一轮中更新输入文件,因为实际原始数据(和R-free flag)并未被修改。
5. 我通过运行AutoSol来获得我现在想要细化的部分模型。我应该将哪个数据文件作为输入:来自HKL2000的原始.sca文件,还是来自AutoSol的overall_best_refine_data.mtz文件?
始终使用AutoSol输出的MTZ文件。它包含一组新的R-free flag用于优化模型;从.sca文件开始将导致生成一组新的flag,which biases R-free。
6. 为什么phenix.refine不会在优化中使用所有数据?
具有非正常值的反射倾向于降低优化引擎的性能。这些是基于几个标准确定的(详见Read 1999),并在每个宏循环开始时过滤掉。您可以通过设置xray_data.remove_outliers = False来防止这种情况。
7. 我应该运行多少个宏循环?
我们建议至少使用五个以确保收敛,但在某些情况下(特别是结构不精细),可能需要更多才能获得最佳结果。(由于速度考虑,默认值为三个宏周期。)
8. 我该如何决定对我的数据使用什么分辨率限制?
尽管人们普遍认为只是为了减少R因子而丢弃可用数据是不可接受的做法。传统标准包括在平均I/sigmaI低于2的分辨率下截断数据,但这可能排除有价值的数据。有关更多详细信息,请参阅有关未合并数据的文档(尤其是引用的参考文献)。通常,在最后相位的细化中包括额外的较弱数据可能是有用的,因为通过最大似然优化执行的加权防止这些降低模型,并且在某些情况下可以改善它。(请注意,更多数据也会导致phenix.refine运行时间更长,这通常会对分辨率产生实际限制。)
9. 是否可以针对已经通过各向异性缩放或椭球截断修改的数据进行优化?
没有技术理由说明这是不可能的。但是,应该避免修改数据,原因如下:
1. 我什么时候应该使用模拟退火?
当模型远离收敛时,模拟退火(SA)在优化的早期是最有用的。手动建立的模型或涉及显着局部构象变化的MR解决方案是常见的输入,其中SA可以通过简单的梯度驱动优化来改进。稍后在细化和/或高分辨率下通常没用。
2. 我什么时候应该使用刚体优化?
这通常只需要在分子置换后执行一次,除非您稍后dock其他结构域。在以后的运行中继续使用刚体优化不会改善您的结构,只会添加运行时间。
3. Old fix_rotamers选项发生了什么变化?
从版本1.8.3开始,真实空间优化策略现在包含全局最小化和局部拟合;后者类似于fix_rotamers,但速度更快,并且具有骨架灵活性。请注意,它不会在非常高或低分辨率或者当存在明确的氢原子时运行。
4. ordered solvent method何时有用?
根据数据质量不同,这可以达到约2.8 Å的低分辨率极限。在原子分辨率(超过大约1.2 Å)的情况下,在没有放置水的情况下,它在优化的早期是有用的,但随着结构变得更加完整,它可以去除较弱的,部分占用的水,它无法处理静态无序(替代构象)。
1. 我应该何时优化几何和/或B因子约束权重?
如果自动加权没有为X射线和约束项选择好的缩放,这可能是有益的; 这通常可以通过高于预期的键和角度RMSD来识别。一般来说,优化权重很少会产生损害,并且通常会产生明显更好的优化,但除非您拥有高度并行的系统,否则它比普通优化要慢几倍。但是,我们强烈建议在最后一轮改进中进行权重优化,这对防止过度拟合至关重要。
2. 如何手动设置目标权重?
我们通常的回答是:不要手动执行此操作,而是使用自动优化。虽然这需要花费更长的时间才能运行,但实际上大多数用户将花费相同的时间通过反复试验手动调整权重。如果您确定需要手动控制,则参数fix_wxc(对于几何约束)和fix_wxu(对于B因子约束)将设置权重。
3. 为什么即使我选择了权重选项,phenix.refine也不会使用权重优化?
因为权重优化会考虑(并试图限制)R-work和R-free之间的差异,如果这些统计数据已经非常接近(或者如果R-free大于R-work),它将自动禁用,如经常在MR解决方案的优化开始时发生。
4. 我的衍射率是X埃; RMS(bonds)和RMS(angles)应该是多少?
这有点争议,但高分辨率的精细蛋白质结构的绝对上限对于RMS(键)通常为0.02,对于RMS(角度)为2.0;通常它们会很低。随着分辨率的降低,几何约束的可接受偏差也会减小,因此在3.5埃时,更合适的值将是0.01和1.0。我们建议在validation summary使用POLYGON tool,以相似的分辨率判断您的结构相对于其他人的结构。
5. 为什么我的输出模型的几何(RMS(bonds)和RMS(angles))很差?
这通常意味着自动X射线/几何加权不能正常工作;如果起始模型的RMS几何也很差,有时会发生这种情况。优化权重(optimize_xyz_weight =True,或“优化设置”选项卡中的等效GUI控件)通常可以解决此问题。
6. 权重优化后,输出模型中的RMS(bonds)和RMS(angles)太低-我怎样才能使它们更高?
这是关于几何偏差的常见误解,部分基于其他改进程序的轶事经验。目标RMSD来自于非常精确的高分辨率小分子结构,因此它们反映了几何中应该发生的真实变化。在较低分辨率下,即使您知道键长和键角必须与高分辨率结构中的变化一样多,但没有足够的实验数据来说明它们应该偏离预期值的方向。因此,精细RMSD低于目标是合理的。(有趣的是,我们发现phenix.refine通常会使用与其他程序类似的R-frees其优化为更严格的RMSD。这可能反映了在几何约束,X射线目标或优化方法中使用的不同方法,但不应引起关注。)
7. 我有这个结构的实验相位,但起始map很差。我应继续使用MLHL目标吗?
用于约束细化的实验相位描述了每个角度的双模态概率分布,而不是用于生成map的单个值。在大多数情况下,额外的约束不会损害细化,并且往往可以有所帮助。
8. 为什么phenix.refine弄乱我的配体几何?
当使用ReadySet从PDB文件生成约束时,通常会发生这种情况,并且配体代码在化学成分数据库中无法识别。eLBOW将尝试仅基于坐标来猜测分子拓扑,但这是不精确的,可能无法产生所需的结果。为获得最佳结果,应使用SMILES字符串或类似的拓扑信息源在eLBOW中生成对非标准配体的限制。
9. 我该怎么做才能使我的低分辨率结构更好?
通常,如果您的结构中存在NCS,则应始终以低分辨率使用NCS约束;值得尝试Cartesian (global)(笛卡尔(全局))和扭转约束,看看哪种方法最适合您的模型。这一点通常有助于几何和过度拟合,尽管它本身是不足够的。还有几种不同类型的约束专门用于帮助进行低分辨率优化
参考模型:这将扭转角度限制在高分辨率模型中,使用轻微释放不同角度(允许局部变形或结构域运动)。这通常是最佳选择,通常可以改进Rfree,过度拟合和验证统计数据。但是,它取决于合适的参考模型的可用性。
二级结构限制:这些只是蛋白质螺旋和sheet中与核酸碱基对氢键原子之间的谐波距离限制。注释可以是自动的,也可以是手动的。目前的主要局限是需要反常值过滤,这可以消除虚假bonds,但也可以消除一些真正的bonds。它对验证统计数据的影响很小,通常在R-free上没有影响,但它确实保持了二级结构的稳定性。
10. 我的离子非常接近水分子/蛋白质原子,而phenix.refine一直保持着将它们分开。我怎么能阻止这种行为?
使用phenix.ready_set或phenix.metal_coordination生成自定义键(以及可选的键角)约束,这些约束将输出到以“.edits”结尾的参数文件。如果您使用的是PHENIX GUI,那么在phenix.refine界面中有一个ReadySet工具栏按钮,它会自动加载输出文件以便在phenix.refine中使用。
11. 我以前在PHENIX GUI中使用ReadySet生成了自定义约束,但原子改变了。现在phenix.refine因为无法找到原子选择而崩溃了,应如何删除旧的自定义限制?
在“实用工具”菜单中,选择“清除自定义限制”。
12. “sigma”对于几何约束意味着什么,什么值是合适的?
sigma是目标值的估计标准偏差(esd)。对于distance(bond)限制,sigma将以Å为单位;对于键角和二面角限制,它将以度为单位。典型的共价键将具有0.02或0.03的sigma,并且较弱的“键”限制(例如氢键或金属配位限制)将更宽松,其中sigma在0.05和0.1之间。对于键角限制,sigma通常为几度;对于二面角,高达20~30度并不罕见。
13. 我希望我的配体几何绝对完美,不会偏离目标值。我可以将sigma设置为零或极低值吗?
不能将sigma设置为零,因为约束上的权重等于1/sigma^2。非常低的值不会崩溃,但它几乎肯定会混淆最小值并导致次优结构,因为这些约束将主导目标和梯度,迫使最小值采取不合适的大步骤。
14. 我的结构的一部分特别差; 如何才能使几何约束更严格,只针对那些原子?
参数scope refinement.geometry_restraints.edits.scale_restraints允许您对指定原子选择的限制进行加权,适用于键长,键角,二面角和手性的任意组合。例如:
refinement. geometry_restraints. edits. scale_restraints {
atom_selection = "chain B"
scale = 2.5
apply_to = *bond *angle dihedral chirality }
您可以指定多个此类参数块,通常小于10个(如果你愿意,你也可以减轻权重)。请注意,由于这会影响最小值的路径,因此总体几何RMSD(以及未缩放的约束偏差)可能会因此而改变。
15. 我可以使phenix.refine约束RNA / DNA碱基对的平面性吗?
目前,这需要单独指定每个碱基对作为自定义平面度约束(参数范围refinement.geometry_restraints.edits的一部分),这对于大型结构来说可能过度耗时。自动平面性限制可能会增加在未来的版本。
16. 我可以使用参考模型来约束配体坐标吗?
参考模型约束只适用于与大分子。但是,您可以对任何原子子集使用单独的谐波约束; 这些将选定的原子束缚到它们的初始坐标。当你已经具有良好的几何形状和适合受约束原子的映射时,这可能是有效的; 但是,它不允许真正的构象差异。
17. 如何阻止模拟退火将某些原子推离密度太远?
谐波约束适用于这种情况。这在生成模拟退火省略图时尤其有用,其中原子可以移动以填充省略的散射体留下的空隙。
1. 我什么时候应该使用非晶体对称(NCS)约束?
这当然要求NCS实际存在于晶体中。NCS限制的近似截止值为2.0埃-在更高的分辨率下,单独的数据通常就足够了,但是在较低的分辨率下,通常需要额外的限制。
2. 全局和扭转NCS之间有什么区别,我应该选择哪一个?
全局NCS限制组作为刚体,其中每组中的所有原子预期通过单个旋转和平移操作与其他原子相关。这不涉及相关分子中的局部变形,即使在较低分辨率下也是常见的。扭转NCS限制了约束二面角,并且如果真正不同则允许它们不受约束。如果激活NCS约束,则此选项已成为默认选项,因为它通常会导致明显更好的优化,并且很少比完全不使用约束更糟糕。
3. 如何从AutoSol或phenix.find_ncs指定.ncs_spec文件以用于优化?
包含旋转和平移矩阵的.ncs_spec文件仅用于密度修改和模型构建。为了改进,NCS关系总是作为原子选择给出,并且在默认扭转NCS约束的情况下,自动检测到的约束组应非常准确。
4. 如何在Phenix GUI中手动定义NCS组?
如果使用扭转NCS约束,则不需要这样做,但您可以通过从“实用程序”菜单中选择“检测NCS组”手动输入原子选择。如果您已经确定了一个结构,其中需要手动分组进行优化以在低分辨率下正常运行,这可能表示phenix.refine中存在bug; 请通过电子邮件“bugs@phenix-online.org”直接与我们联系。
对于全球/笛卡尔NCS限制,手动选择往往是 必要的,如果选择了全局选项相同的菜单项将打开一个不同的选择窗口。但是,我们建议您在决定使用全局参数化之前先尝试扭转NCS约束。
5. 两种NCS约束类型都会使我的结构变得更糟-我该怎么办?
如果你要以高分辨率(优于2.0埃)优化结构,这并不意外;在这种情况下,通常不需要NCS限制。在较低的分辨率下,这可能表示存在bug;我们鼓励您直接与我们联系。
6. NCS约束对电子密度图有何影响?
这些密度图只会受到与NCS相关的链相对相似的影响,因此map相位将反映这种相似性。但是,不会使用NCS关系直接修改map。如果您希望phenix.refine在map上执行NCS平均,则可以选择单个map系数。
7. NCS限制是否适用于B因子?
默认情况下不适用; 我们的测试表明这很少有益,并且在相当一部分结构中它实际上可能产生更差的结果(由于NCS相关拷贝中存在的无序性的真实差异)。如果希望在NCS组之间限制B因子,请在“NCS选项”窗口中选中标记为“Restrain NCS-related B-factors”的框,或在命令行上指定ncs.restrain_b_factors = True。
1. 我什么时候应该使用TLS?
TLS优化通常在任何分辨率下都有效;在低分辨率下,最好将每个链组成一个组,而不是试图将它们分成更小的组。但是,最好等到优化结束时添加TLS;在此之前,您应该仅使用各向同性ADP进行优化。
2. 我可以同时使用TLS和各向异性ADP吗?
是的,但不是相同的原子-因为TLS本质上是受约束的各向异性优化,这两种方法是互斥的。
3. 各向异性与各向同性B因子/ADP的开关在哪里?
phenix.refine没有用于定义ADP参数化的单个全局开关;相反,当定义“Individual ADPs”策略时,该程序使用几个标准来确定应如何处理原子:
默认情况下,如果分辨率至少为1.7埃,输入模型中各向异性的原子(即具有ANISOU记录)将保持各向异性; 各向同性原子保持各向同性。
如果分辨率较低,除非另有说明,否则所有原子都将转换为各向同性。
各向同性和各向异性原子的原子选择也可以明确定义。通常,氢将始终是各向同性的;蛋白质原子在高分辨率下是各向异性的,有时也是水。
在GUI中,在对话框中预定义了几个常用参数,用于输入ADP选择。请注意,虽然可以在一次运行中组合所有不同的ADP优化策略,但是单个和分组优化的原子选择可能不重叠,也不能选择各向异性ADP和TLS组。
4. 我何时应该改进各向异性ADP而不是TLS组?
您应该打开各向异性ADP没有精确的截止值,但这些是近似准则,假设数据实际上已完成指示的分辨率:
在1.4埃或更高的分辨率下,各向异性地优化蛋白质/核酸/配体重原子(C/N/O或更重),但采用各向同性的水。
在1.1埃或更高时,水也应是各向异性的。
各向异性优化氢几乎从来都不合适。
可能存在允许在略低的分辨率下进行各向异性优化的情况,但1.7埃可能是下限。有时可能会对金属离子例外,因为它们散射非常强烈。与往常一样,您应该使用R-free下降来判断参数化的变化是否合适 - 减少0.5%(即0.005)或更好表示成功。
5. 我什么时候应该优化grouped B-factors/ADPs而不是individual?
由于它取决于晶体的几种性质,包括分辨率,溶剂含量,NCS的存在等,因此难以给出准确的规则。通常,数据与参数比越高,单个ADP越可能是工作良好。作为一个近似的例子,考虑这两个假设结构:
每个不对称单元的单个蛋白质链的3.5埃结构,溶剂含量为38%。
具有3个NCS相关链的4.0埃结构,具有70%的溶剂含量。
在这种情况下,后一种结构可能可以用单独的ADP进行优化,而前者则更为边缘化。如果有疑问,可以使用分组的ADP完成早期的优化,当结构接近收敛时切换到个体。一般来说,在某些时候通常值得尝试单独的ADP; 最终对R因子的影响(主要是R-free,和R-work和R-free之间的差距)是最重要的指导原则。
1. 我什么时候应该使用孪晶优化?
只有当Xtriage指示强度统计信息反常且无法通过选择错误的空间组来解释时,才应执行此操作。在Xtriage中显示的估计的孪生规则特异性孪生分数不应用于确定是否存在孪生。
2. 我进行了双重改进,我的R-free下降了1%; 这是否意味着我的结构是孪生?
不,因为在有和没有孪晶的情况下计算的R因子不一定是相同的。在phenix.model_vs_data中,如果应用双π定律将R-work减少至少2%,则该结构仅被视为孪生。请注意,如果指定twin_law= Auto,则phenix.refine将使用相同的过程来确定孪生法(如果有)。
3. 但是,如果报告的孪晶分数接近50%怎么办?
这通常意味着您的数据合并不足,即晶体对称性太低,并且应用的孪生算子正在取代对称操作。如果是这种情况,您应该将数据合并到更高的对称性,并将模型减少到新的ASU。当然也有可能有完美的孪晶,但是由 phenix.refine 产生的高孪晶分数本身并不表示孪晶。
4. 我的数据有多个孪晶法则;我可以在Phenix中使用它们吗?
目前我们只支持单一孪生法则;能够精炼四面体孪晶结构的程序是REFMAC和SHELXL。
5. 孪生优化的缺点是什么?
具体而言,在Phenix中,孪晶优化使用最小二乘(LS)目标而不是用于传统优化的更强大的最大似然目标。此外,孪晶优化不会使用实验相位(如果可用)作为限制。一些改进协议可能不适用于结对,尽管截至2013年4月,其中大部分已经修复。更一般地,输出映射系数将具有比传统映射明显更差的模型偏差问题; 随着孪生分数接近0.5,这种效应会增加。
1. phenix.refine因错误中止,提示“R-free flags not compatible with F-obs array: missing flag for 100 F-obs selected for refinement”,我该如何解决?
这意味着实验数据数组包含R-free flag数组中不存在的反射(即使标志为False,这也是必需的)。使用反射文件编辑器扩展现有的R-free标志以覆盖文件中的所有反射。确保选中标记为“Extend existing R-free array(s) to full resolution range”的复选框。
2. phenix.refine停止,并显示有关模型正在针对不同的R-free标志集进行优化的错误消息。我怎样才能解决这个问题?
首先,确保您没有实际生成一组新的R-free标志;一旦给定数据集的这些标志,就应该在整个构建和优化过程中继续使用它们。错误消息旨在防止意外发生这种情况。但是,如果您已收集新的更高分辨率数据并扩展旧的R-free标志,则可能会忽略错误消息。R-free标志比较基于存储在输入PDB文件中的REMARK记录中的信息,因此如果您编辑PDB文件并删除包含单词“hexdigest”的行,则细化将能够继续。
3. 我有一个模型,以前针对我无法访问的前一组R-free标志进行了改进。当我改进这个模型时,如何避免偏置R-free?
有几种方法,但最简单的方法是重置B因子(使用PDBTools或phenix.refine中的“Modify start model”选项)并在坐标上运行模拟退火。如果您特别担心偏差,您可以交替随机化坐标并执行能量最小化,或者从原始模型计算的相位开始构建一个全新的模型。但是,我们通常发现退火足以消除原始无R标志的任何“memory”。
4. 我的分辨率是X埃,我的R/R-free是Y和Z。我做过优化吗?
通过查看POLYGON可以获得部分答案,POLYGON绘制了在相似分辨率下求解的PDB结构的统计直方图,并将这些与输出模型的统计数据进行比较。作为一般规则,不应仅使用R因子来确定结构是否“完成”,而应结合验证报告进行检查。
5. 我的分辨率是X埃,结构完整且经过充分验证,map看起来很好,但我的R和R-free仍然非常高。我怎样才能降低它们?
对此有几种可能的解释:
具有相对较小的孪生分数(可能是10%)的孪生可能不会明显影响密度图质量,但仍然可以对R因子产生显着影响。运行Xtriage寻找孪晶的证据,以及在phenix.refine中使用的可能的孪晶定律。(如上所述,一次只能使用一项孪生法则。)
未确诊的平移NCS(AKA平移假对称)可以产生类似的效果。在这种情况下,衍射图像可能已经被错误地处理,因此错过了由tNCS导致的较暗点。可以在原始图像上运行程序LABELIT,或者更具体地命令labelit.index(与PHENIX一起分发,但在GUI中可用)以提供适当的索引参数。
6. R-work和R-free之间的差距非常大 - 我该如何解决这个问题?
通过增加更多约束和/或收紧标准几何约束,通常有助于精修过度拟合。如果输出几何已经在合理的范围内(通常RMS(bonds)<0.016和RMS(angles)<1.8),尝试包括:如果存在NCS则添加NCS限制,二级结构限制或参考模型限制(如果高分辨率结构可用)。在较低的分辨率(低于3.0埃),谨慎的尝试分组ADP细化,如果绝望,尝试Ramachandran限制。TLS细化通常可以改善各种分辨率的过度拟合。但是,根据过度拟合的程度,可能需要首先进行大量的手动重建。(注意,如果在先前没有过度拟合的模型的细化之后突然出现大的R-free / R间隙,则这通常表示细化的参数化不正确,例如以不适当的分辨率使用各向异性ADP。)
7. 我进行了一轮细化,在coot重建,并再次细化。我之前的R-free是0.25,但新的细化开始于0.35。它为什么这么高?
由phenix.refine报告的初始R因子没有大量溶剂校正,这通常对R因子具有显着影响。一旦执行该步骤(在第一个宏周期的开始),R-free应立即下降到大约预期值。
8. 为什么phenix.refine给我一个与程序X不同的R因子?
对此有很多解释。即使没有最小化,单独的本体溶剂和缩放方法可以在计算的R因子中占据多达1%或更多的差异。对于细化结果,目标函数,约束和最小化器的差异可能更大。在某些情况下,解释就像为一个或另一个程序运行太少的细化周期一样简单。一般来说,如果您发现phenix.refine的表现比其他程序差得多,我们建议您通过bugs@phenix-online.org与我们联系。
9. 用于实空间细化的map中是否包含R-free标记的反射?
不,这几乎可以保证没有R-free;在map计算之前,内部会删除这些反射。但是,除非您明确要求,否则输出映射将包含这些反射。
这经常表明MR放置了太少的结构拷贝,并且需要添加额外的链。请记住,基于马修斯系数(例如,由Xtriage执行)的晶胞内容的预测 仅提供估计而不是确切的答案。在高分辨率下,溶剂含量为40%或更低是非常常见的。
2. 为什么我在疏水性空隙中看到差异(mFo-DFc)图中的负斑点?
在以前版本的Phenix中,本体溶剂掩模通常被扩展到包括这些区域。这应该不再是2013年7月的问题,但如果您继续看到这种影响,请通过bugs@phenix-online.org与我们联系 。
3. 经过坐标和B因子细化后,我结构中的重原子具有负mFo-DFc峰。我怎么去除这些?
改善占用率通常可以解决这个问题。或者,如果预期在用于数据收集的波长上存在大量的反常散射,则反常组的细化也可能是有帮助的。我们不建议手动设置B因子并关闭有问题原子的细化。
4. 在细化之后,mFo-DFc图具有正确放置的原子周围的正密度,这些原子已经处于完全占用状态。我的模特缺少什么吗?
通常这意味着输入模型的初始B因子太高而无法收敛。通常,最小化器非常擅长将低B因子提高到正确值,但在相反方向则不是。如果您从较低分辨率的模型开始细化,或者如果您在Coot中构建新的残基并且默认的B因子远高于它们应该的值(通常仅在原子分辨率下发生),则可能发生这种结果。要解决此问题,您只需将B因子重置为适当的低值即可。这可以在细化开始时通过设置参数refinement.modify_start_model.adp.set_b_iso来完成;在GUI中,可以在“修改启动模型”->“修改ADP..”下的“设置”菜单中找到它。
5. 为什么Phenix验证报告的Ramachandran异常值和Coot / Procheck / PDB /其他程序不同?
Phenix中使用的phi/psi分布与MolProbity服务器中的phi/psi分布相同(Chen等人,2010),并且基于8000个高分辨率晶体结构。现在不同残基类别有六种的分布(一般,甘氨酸,Ile/Val,pre-Pro,顺式Pro和反式Pro)。这些分布以2度为增量存储。其他程序通常使用较旧和/或较不精确的分布来评分phi/psi角度,这经常导致对图的允许区域和异常区域的边界上的残基的不一致。我们建议您主要依靠Phenix(或MolProbity)中的结果,因为我们使用的分布非常准确并且基于最新的结构数据。
6. 在运行溶剂更新后,为什么验证结果中会出现一堆与水分子的冲突?
phenix.refine在将溶剂原子置于未建模密度时相对具有侵略性。然而,如果密度代表离子,未建模的残基或替代构象而不是溶剂,这有时可能导致冲突。出于这个原因,我们建议在任何冲突的水原子被移除后最后一轮改进不包括溶剂更新(无论分辨率多少)。(如果可能的话,您还应该尝试对观察到的密度特征进行建模,尽管这并不总是很简单。)
7. 当水原子处于正mFo-DFc峰值时,它意味着什么?
这表明水实际上更重。如果密度形状(对于2mFo-DFc和mFo-DFc图)仍然是相对孤立和球形的,这表明是某种离子(氯离子,钙等)。您可以使用phenix.refine中的内置离子识别来尝试对其进行建模,或者根据对当地环境的检查手动进行分配。我们还建议查看反常差异图,因为许多离子在典型的数据采集波长下会有明显的异常散射。或者,如果密度更狭长,则表明结合了更大的配体。
8. 我还有一些我无法识别的神秘的mFo-DFc斑点; 我该怎么办?
一种选择是使用配体识别工具试图将各种常见的小分子置于密度中; 我们建议在过滤结果时要非常保守。检查纯化,结晶和冷冻保护的条件是非常有帮助的,因为这些通常导致缓冲组分在图中可见。但是,如果您无法正确识别斑点,我们建议将它们留空(也许还可以在提交备注或出版物中注明)。我们不建议用水分子填充它们,因为这会错误地表示点的真实身份,我们也不建议使用“未知”原子作为占位符(特别是因为它们与Phenix中的大多数工具不兼容)。
1. 我什么时候应该用氢细化?
这主要取决于个人偏好。使用明确的氢原子可以在任何分辨率下改善几何形状; 在更高的分辨率,大约2埃或更高,他们通常也会提高R-free。在原子分辨率(1.5 A或更高)下,它们应该始终是最终模型的一部分。请注意,除非您具有真正的亚原子分辨率(0.9 A或更高),否则氢应始终细化为“riding”,这意味着它们的坐标由重原子定义,而不是根据X射线数据单独提炼。
2. 我怎么能告诉phenix.refine在我的模型中添加氢?
命令行程序不添加氢;这是由一个单独的程序phenix.ready_set执行的。但是,在GUI中,“优化设置”选项卡中有一个选项可以添加氢
3. 水分子怎么样?
尽管phenix.ready_set包含向水中添加氢的选项,但除非您具有极高的分辨率或中子数据,否则我们不建议这样做。
4. 当我在Coot中运行真实空间细化时,为什么PHENIX添加的氢原子会爆炸?
0.6.2之前的Coot版本使用具有根据PDB格式版本2标准命名的氢原子的CCP4单体文库的版本;PHENIX可识别这些,但默认为PDB v.3。要协调不同的约定,您可以下载较新版本的单体库并将环境变量COOT_REFMAC_LIB_DIR设置为指向解压缩它的目录。但是,较新版本的Coot似乎没有这个问题。
5. 我用riding氢来细化-为什么它们出现在输出模型中?
phenix.refine将始终输出作为原子模型一部分的所有原子,无论它们在细化过程中如何参数化。使用“riding”模型不一定保证氢位置和B因子是可再现的,因为它们在各种程序(或程序版本)之间不同。
6. 为什么PHENIX不能自动从输出PDB文件中删除氢?
我们强烈反对从模型中去除细化中使用的任何原子,因为它使得复现已公布的R因子非常困难并且消除了关于结构如何被精炼的基本信息。
7. 如何定义XH(氢原子与重原子)键长,为什么它们与分子力学程序中的不同?
由于X射线被电子衍射,因此默认的氢位置被定义为电子云的中心,而不是原子核。在大多数情况下,这导致键长比给予核的距离短0.1。在高分辨率下,这将更准确地模拟X射线数据。用于非健约束的VDW半径将更大,以补偿更短的粘合。
如果您正在进行中子细化,则将使用核位置,从而导致更长的键长和更小的VDW半径。MolProbity验证应该适当地处理两种情况。
1. 我如何模拟带电原子?
电荷在每个ATOM或HETATM记录末尾的第79-80列,紧跟元素符号。格式是电子数+电荷符号,例如“1-”或“2+”。您可以手动编辑PDB文件以添加它,但我们建议使用phenix.pdbtools:
phenix. pdbtools model.pdb charge_selection = “element Mn” charge = 2
这也可以在GUI中“模型工具”下找到。设置电荷的效果是使用修改的散射因子进行X射线细化,如果您注意到离子位置出现差异密度,这可能会有所帮助。请注意,它对几何体没有影响,因为phenix.refine不考虑静电。
2. 我看不到C-gamma原子以外的精氨酸侧链的密度。我应该如何建模呢?
结晶学界的观点在对无序侧链的正确方法上有所不同,对PHENIX和CCP4邮件列表中提到的以下两种方法都有很大的支持:
删除所有密度不可见的原子,但留下残基名称。这可以说是最保守的方法,因为它避免了对数据不支持的任何特征进行建模,并且与缺失loop的处理一致。主要缺点是不美观,因为难以可视化和解释具有缺失侧链的结构的生物效应。轶事证据表明,一些非晶体学家可能会对此感到困惑。
选择一个合适的旋转器,让B因子上升以解决无序。这避免了可能被误认为是其他残基的截断侧链,并且在解释表面静电时更加真实。原子B因子和坐标实际上是针对数据进行细化的,无论多么微弱。它具有潜在的危险性,因为它意味着对这些位置的信任程度高于数据证明的可信度。此外,ADP限制将使附近原子的B因子保持相似(在一定的容差范围内),这通常对于稳定细化是必需的,但可能人为地降低无序侧链的B因子。
第三种方法,将缺失原子的占有率设置为零,但将它们留在模型中,是非常不受欢迎的,因为得到的位置和B因子完全是理论上的(但不是那么明显)。
运行phenix.model_vs_data(或验证GUI)会导致与PDB标头中由phenix.refine报告的R因子略有不同。它们不应该一样吗?
phenix.refine和phenix.model_vs_data使用相同的代码来执行本底溶剂校正和缩放,因此它们应该在给定相同输入的情况下报告大致相同的R因子。从细化中获取PDB文件并通过phenix.model_vs_data运行它时会出现差异。由于PDB格式的精度有限(坐标小数点后三位数,B因子和占用率后两位数),PDB文件中记录的原子属性与实际细化值不完全相同。然而,在实践中,R因子的差异在统计上是不显着的。
3. 我什么时候应该进行反常组的细化?
当您拥有大量强反常散射体时,这是最有用的,其中map伪像很常见。在这种情况下,更精确的原子散射建模也可以改善R因子。对于常规情况通常不是必需的,但是当用于在细化结束时计算反常对数似然梯度(LLG)或残基图时,识别弱的异常散射体(例如来自结晶条件的离子)可能是有利的。
4. phenix.refine如何处理特殊位置的原子?
在特殊位置处理原子有两种方法(例如对称轴):
如果占用率设置为特殊位置的预期值(例如,对于二次轴上的原子为0.5)或更小,则坐标位置将被细化,并且不会与其对称原子相互作用。
如果占用率设置为1,则原子将被约束为保持在特殊位置,并且在计算结构因子时将在内部校正占用率。
请注意,如果使用默认设置,特殊位置上的部分占用原子将定义其占用率; 您可以通过指示phenix.refine从占用细化中删除特定原子选择(关键字refinement.refine.occupancies.remove_selection,或在GUI中编辑占用策略的原子选择)来禁用此功能。
5. 我有多个构象的侧链在对称轴上相互作用,其中'A'构象相互冲突并通过细化移出密度。我怎么能告诉phenix.refine忽略这种交互?
这并不罕见(典型的例子是PDB ID 1GWE中的Tyr378),但它现在不会自动处理,并且PDB格式不提供标记原子的方法以避免冲突。phenix.refine中的解决方案是定义原子选择的参数,应禁用非健限制:
custom_nonbonded_symmetry_exclusions =“链A和resseq 378”
6. 如何从用TLS或各向异性原子细化的结构中提取各向同性B因子当量?
你不需要任何额外的步骤;PDB(或mmCIF)文件中ATOM记录中的B因子列已经是总B因子。
7. 为什么phenix.refine输出ANISOU记录单个原子,即使我只执行各向同性和TLS细化?
TLS细化本质上是受约束的各向异性细化,因此各个原子是各向异性的(只是不是独立的);ANISOU记录只是简单地说明了这一点,因为它们具有标准格式并且被各种程序识别,这与PDB标题中的TLS信息不同。
8. 为什么PDB标题不报告本底溶剂参数K_sol和B_sol?
更新版本的Phenix使用改进的本底溶剂校正和缩放程序,使用完全不同的参数化,我们发现它们表现更好(Afonine等人2013;另见Uson等人(1999)Acta Cryst.D55,1158-1167)。
9. 各种散射表有什么区别?我应该何时使用默认值以外的其他内容?
如果要细化中子结构,当然应该使用中子散射表。其他表格都是X射线特异的;默认值n_gaussian是最好用的,因为它使用动态定义的高斯数来近似表格形式因子,并具有所需的精度。it1992常用于其他程序-这是四个高斯加常数,取自国际表1992版。wk1995来自(Waasmaier&Kirfel 1995),它是五个高斯,比它更准确(但更慢)。
10. 为什么phenix.refine不使用恒定数量的分辨率/为什么phenix.refine不将反射均匀地分配到分辨率箱中?
均匀分档反射不适用于phenix.refine中使用的本体溶剂校正和缩放方法,而是使用对数分级。Afonine等人解释了这一逻辑。(2013年):
“这种方案允许较高分辨率的分区包含比低分辨率分区更多的反射和在更低分辨率有更详细分区而不增加分区总数。使用对数分级的另一个原因是缩放对分辨率的依赖性大约是指数(参见前面的部分),当使用对数分级算法时,这使得缩放因子之间的变化更加均匀。”
1. 我该如何引用phenix.refine?
(Afonine等人,2012年)或(Adams等人,2010年)都是合适的;如果您使用Phenix的其他组件,我们建议使用后者。如果您在GUI中使用了集成的MolProbity验证,您还应该引用(Chen et al.2010)和/或(Echols等人,2012)。
2. 我应该如何在PDB提交中指定细化程序?
如果您存入的PDB或mmCIF文件是由phenix.refine输出的,则标题中已指定了细化程序(包括版本号)。否则,“PHENIX(版本1.8.4)”是合适的(替换为实际版本号)。但是,请注意,如果在结构确定过程中使用了多个细化程序(例如REFMAC和PHENIX),则通常只会在标题中命名最后一个,因此我们建议您在提交期间编辑此信息以完成列表。
3. phenix.refine中使用的基础方法有哪些参考?
对于技术背景,最彻底的来源是(Afonine等人,2012),其中包含所有相关的引用。我们建议所有使用phenix.refine的人在某些时候阅读本文,即使你不关心理论,因为它提供了比本文档更详细的解释方法和动机背后的方法。
4. 我在哪里可以阅读更多关于一般细化原理的内容?
Bernhard Rupp的“生物分子晶体学”是最现代和最完整的参考,包括phenix.refine和许多其他程序中使用的最大似然法的详细解释。
5.13.5 Pymol 基本操作
简介(INTRODUCTION)
Pymol是一个被广泛使用的显示分子结构,给结构作图的软件。还可以做一些简单的测量,修改结构,移动等。此外,它还能做动画。常用操作系统支持,教育版的不能输出高清图。
How to move stuff around in PyMOL? 如何使用鼠标移动分子。
To move molecules around one have to activate 3-button editing mouse mode. Easiest way to do it is to click on "Mouse Mode - 3-Button Viewing" in the lower right corner of the main window. Then you will be able to move single molecule while holding down shift key:
left mouse button rotates molecule
middle mouse button moves molecule
right mouse button moves molecule closer or farther
在右键选中一个molecule/chain/object后,选择drag, 可以同时按下shift只对这一object进行移动。
基本命令(BASIC COMMANDS)(也可使用鼠标操作,但不如命令来得简单)
PyMOL> pwd # show current directory
PyMOL> dir # list file in the current directory
PyMOL> cd directory #change directory
PyMOL> load xxx #注意要打上扩展名,例如1 LMK.pdb,否则会error
PyMOL> creat name, (selection) #name=object to create, selection=atom to include in the new object, 非常实用的功能,创建一个新的物体
Manipulating objects:
PyMOL> show representation, (selection)
PyMOL> hide representation, (selection)
#the available representations are: (前面是个人比较常用的)lines, spheres, ribbon, cartoon, sticks, surface, /// mesh, dots, labels, extent
例如:
PyMOL> stick mol1 & resi 100 # mol1为显示在pymol右边的分子名称
Turn an object on and off:(这个直接用鼠标就ok了)
PyMOL> enable/disable, object-name #turn on/off all representation
Basic atom selections:
name <atom names> 缩写 n.<> 例子: Show cartoon, (n. 1 LDK/)
resn<residue types> r.<>
resi<residue number> i.<>
chain<chain ID> c.<>
elem<element symbol> e.<>
Selection algebra: 就是怎么选择
交集是and、&,例如 1 LDK and chain A and i. 1-103
并集是or/|,补集是not/!
Change your point of view
PyMOL> zoom, (selection) #fit the selection to screen
PyMOL> orient, (selection) #align molecular axisc
PyMOL> enter, (selection) # size not changed
PyMOL> turn axis, angle #rotate camera
PyMOL> move axis, distance #translate camera
Align (很重要的功能):
align (source), (target) #the source object will be moved and rotated to fit the target object
例如: align (prot1 and chain A), (prot2 and chain B) 按整条链叠合
PyMOL> align mol1 & resi N1, mol2 & resi N2 按某一个残基叠合
PyMOL> align mol1 & resi N1-N2 & name n+ca+c+o,mol2 & resi N3-N4 & name n+ca+c+o 按某段残基的主链进行叠合
在align的过程中会产生一个root mean square deviation (RMSD),这个值可在一定程度上衡量alignment的效果。
Set control (可以通过这个命令来调整所有参数的值,非常有用)
PyMOL> set sphere_scale, 0.5, (n. Fe) #decrease Fe atom size to 0.5
PyMOL> set bg_rgb, [1,1,1] #set background as white
PyMOL> set ribbon_sampling, 1
Alter (可以用来改变结构,比如显示的二级结构定义,改变chain ID, 改变序号等)。
PyMOL>alter (chain A),chain='B'
PyMOL>alter (all),resi=str(int(resi)+100)
PyMOL>sort
PyMOL>alter (name P), vdw=1.90
PyMOL>Rebuild
PyMOL>Alter resi 1~15 and chain A, ss=’h’
PyMOL>Rebuild
Cartoon display
显示cartoon时候可以有几个选项。
PyMOL> set cartoon_fancy_helices, 0 # 显示默认圆的螺旋
PyMOL> set cartoon_fancy_helices, 1 #显示扁的螺旋
PyMOL> set cartoon_side_chain_helper, off #在cartoon上显示侧链的时候,主链原子也显示
PyMOL> set cartoon_side_chain_helper, on #在cartoon上显示侧链的时候,只显示侧链
Display Surface
显示分子表面的选项。
PyMOL> set surface_quality, 0
PyMOL> set surface_quality, 2
PyMOL> set solvent_radius, 1.0
PyMOL> set solvent_radius, 2.0
PyMOL>set mesh_color, grey80
Display nucleic acids in cartoon
显示核酸时的选项:
PyMOL> set cartoon_ring_mode, 3
PyMOL> set cartoon_ring_finder, 1
PyMOL> set cartoon_ladder_mode, 1
PyMOL> set cartoon_nucleic_acid_mode, 4
PyMOL> set cartoon_ring_transparency, 0.5
Measurement
(又一个十分重要的功能,可以真正挖掘结构的意义)
PyMOL> dist #测量两个原子之间的距离,ctrl+右键选择第一个原子,ctrl+中键选择第二个原子,然后测量dist。
PyMOL> angel # 测三个原子之间的夹角, ctrl+中键选择第三个原子,然后测量angle
PyMOL> dihedral #测量四个原子之间的二面角
Coloring Molecules
To assign helices, sheets and loops individual colors, do:
PyMOL> color red, ss h
PyMOL> color yellow, ss s
PyMOL> color green, ss l
When the colour bleeds from the ends of helices and sheets into loops, do:
PyMOL> set cartoon_discrete_colors, 1
B-factor coloring can be done with the spectrum command. Example:
PyMOL> spectrum b, blue_white_red, minimum=20, maximum=50
PyMOL> as cartoon
PyMOL> cartoon putty
Coloring insides and outsides of helices differently
这一功能很有用,可以把helices里面和外面显示不同颜色
The inside of helices can be addressed with:
PyMOL> set cartoon_highlight_color, red
Isomesh
Isomesh can be used to display electron density map. Isomesh可以用来显示电子云密度图。Pymol can read CCP4 maps.
PyMOL> load ref.pdb
PyMOL> load 2fofc.ccp4
PyMOL> isomesh full_map, 2fofc, 1.0 ; # display density mesh for the entire map at 1.0 isosurface level
PyMOL> isomesh brick_region, 2fofc, 1.0, ref//C/26/, 3.0 ;
# parallelepiped with 3.0 Å atom buffer
PyMOL> isomesh carve_region, 2fofc, 1.0, ref//C/26/, carve=2.1 ; # only show density within 2.1 Å of atoms
SYMEXP
Symmetry expansion is accomplished using “symexp” command, which relies upon CRYST records present in the input PDB file. 这一功能用来显示对称性分子。
PyMOL> symexp prefix, object, selection, cutoff # example:
PyMOL> symexp sym, ref, ref, 5
Ray
PyMOL> ray 576,576 # 8inch * 72dpi
PyMOL> ray 800,800 # 8inch * 100dpi; or a 4inch * 200 DPI photo; or 1x800.
PyMOL> ray 2400, 2400 # 8inch * 300dpi; 6"x400dpi, etc...
PyMOL> png fig2b, dpi=300
PyMOL> get_view # to get the current view, so that you can set next time.
PyMOL>set_view
Example:
PyMOL> color violet, map # sets map to violet
PyMOL> set mesh_width, 0.5 # makes meshes thinner for ray-tracing
PyMOL>set mesh_color, grey80
PyMOL> bg_color white #sets background to white
PyMOL> set ray_trace_fog, 0 # turns off raytrace fog--optional
PyMOL> set depth_cue, 0 # turns off depth cueing--optional
PyMOL> set ray_shadows, off # turns off ray-tracing shadows
PyMOL> ray 1024,1024 # this would create a 1024x1024 pixel ray-traced image
PyMOL> png image.png # output final image
PyMOL> isomesh map_name, map, 2.0, ligand, carve=1.6 # set contour level 2.0. Carve 1.6 Å around the ligand
PyMOL> set mesh_width, 0.5
PyMOL> set mesh_radius, 0.02
PyMOL> set mesh
PyMOL> set dash_gap, 0.5
PyMOL> set dash_radius, 0.05
PyMOL> set stick_radius=0.2
PyMOL> alter all, vdw=0.3
PyMOL>rebuild
PyMOL> set stick_transparency, 0.5, object
PyMOL> set sphere_transparency, 0.5, object
PyMOL> color grey80, map_name
PyMOL> Viewport 800,800
PyMOL> ray 2400, 2400
PyMOL> png fig_name
Pymol的功能很强大,更多指令可以参考pymol wiki http://pymolwiki.org/index.php/Main_Page
(2018年 金腾川 撰)
5.13.6 用Pymol创建动画
简介(INTRODUCTION)
任何分子可视化程序都允许用户将结构的各个视图保存到一组图像文件中。然后可以将这些图像文件转换为一个movie文件。使用PyMOL可以轻松执行简单的任务。例如,创建旋转或摇摆分子的movie。
实验步骤(PROCEDURE)
1. 创建帧(.png图片)
2. 在pymol中调整分子的方向使得螺旋是垂直的
3. 定义帧,输入> mset 1,180 (#预定义180个电影帧)
4. 定义动作(旋转)
输入> util.mroll 1, 180, 1(# 180度旋转,产生1度间隔的180帧。)
5. 图像润色
输入>set ray_trace_frames, 1(# 光线追踪,使帧图像更光滑)
6. 设置缓存
输入>set cache_frames,0(#
缓存)
7. 保存movie
输入>mpng frame(# 保存帧为.png文件,文件名称以frame开头。Windows系统默认保存至桌面)
8. 图像重组
通过图像转换软件将.png转换成.jpg。这里我采用“Gif Movie Gear”软件由创建好的这些.png文件创建movie。
5.13.7 用pymol和APBS画静电表面
基本流程
1. 运行pdb2pqr将pdb文件转化成pqr文件同时生成apbs的输入文件($ pdb2pqr --ff parse --apbs-input *.pdb *.pqr)(力场可换成其他的,如 amber, charmm等)。
2. 运行apbs进行泊松-波尔兹曼方程计算,输出*.dx($ apbs *.in)。
3. 可视化
网上有服务器(PDB2PQR Server)供使用,可进行前两步,其使用的pdb2pqr的版本有2.1.0和2.1.1两种,apbs的版本为1.3。
在pymol中的APBS Tools集成了上述步骤:
4. 在pymol中打开一个*.pdb
5. 打开Plugin>APBS Tools2.1...
6. 打开Program Locations程序会自动在$PATH中搜索apbs, psize.py, pdb2pqr.py,如果未找到需手动输入路径。(psize.py不是必需的,位于apbs安装目录下的/share/apbs/tools/manip/)(pdb2pqr.py只在pqb2pqr1.8及之前的版本中有,新版本改为pqb2pqr)
7. 点击Set grid,程序将在后台运行pdb2pqr,之后可以在Configuration中修改参数(注意温度的默认值为310)。
8. 点击Run APBS,程序将在后台运行apbs。
9. 一般对图像进行如下修改。
10.在Visualization中的Molecular Surface部分,Low调为-5,High调为5,点击Update。
11. PyMol>set surface_quality,2
PyMol>set solvent_radius,0.5
注:
已有pqr文件可选择不使用pdb2pqr.pqr,在第三步不必输入pdb2pqr.py的位置,在Main中点Choose Eternally Generated PQR,选择已生成的*.pqr,再执行第四步。
已有*.pdb/*.pqr和*.dx,均用pymol打开(若文件名相同直接用load命令后一个文件会把前一个文件覆盖)打开Plugin>APBS Tools2.1...直接跳到第六步。
使用APBS Tools与使用网上的服务器得到的结果会不同,使用不同版本的apbs结果也会有差异。
5.13.8 用Autodock做分子对接
简介(INTRODUCTION)
要求:已经安装好AutoDockTools,AutoGrid,AutoDock并正确配置了环境变量。
受体为刚性则跳过括号里的步骤,受体某些残基为柔性则要执行括号里的步骤。
实验步骤(PROCEDURE)
1. 准备工作
准备好受体与配体的pdb文件(为了之后方便最好复制到工作目录)。
进入工作目录。($ cd)
打开AutoDockTools。($ adt)
2. 处理受体的pdb文件(去水加氢)
File>Read Molecule 选择受体的pdb文件,点击Open。
Select>Select From String 在Residue框中输入HOH*,Atom框中输入*,点击Add,点击Dismiss。
Edit>Delete>Delete Selected Atoms 点击CONTINUE。
>Hydrogens>Add 点击OK。
取消显示受体以方便下一步观察配体。
(Flexible Residues>Input>Choose Macromolecule... 选择受体,点击Select Molecule,点击Yes,点击OK。)
(Select>Select From String 在Residue框中输入残基,例如ARG8,点击Add,点击Dismiss。)
(Flexible Residues>Choose Torsions in Currently Selected Residues...)
(Flexible Residues>Output>Save Flexible PDBQT...)
(Flexible Residues>Output>Save Rigid PDBQT...)
3. 处理配体的pdb文件
Ligand>Input>Open... Files of type选PDB files 选择配体的pdb文件,点击Open,点击OK。
Ligand>Torsion Tree>Detect Root...
Ligand>Torsion Tree>Set Number of Torsions... 选择fewest atoms,填一个尽量大数字,点击Dismiss。
Ligand>Torsion Tree>Choose Torsions 设置可以旋转的键,一般使用默认的即可。
Ligand>Output>Save as PDBQT... 点击Save。
4. 设置搜索空间
Grid>Macromolecule>Choose... 选择受体,点击Select Molecule,点击OK,点击Save。
Grid>Grid Box... 调整参数number of points in ?-dimension和? center使结合位点位于格子中。 File>Close saving current
5. 准备AutoGrid参数文件
Grid>Set Map Types>Choose Ligand... 选择配体,点击Select Ligand。
(Grid>Set Map Types>Choose FlexRes... 选择受体,点击Select molecule providing flexible residues。)
Grid>Output>Save GPF... 点击Save。
6. 运行AutoGrid
Run>Run AutoGrid... 点击Parameter Filename后的Browse,选择刚才生成的gpf文件,点击Open,点击Launch。等待程序执行结束。
7. 准备AutoDock参数文件
Docking>Macromolecule>Set Rigid Filename... 选择受体的pdbqt文件,点击Open。
(Docking>Macromolecule>Set Flexible Residues Filename...)
Docking>Ligand>Choose... 选择配体,点击Select Ligand,点击Accept。
Docking>Search Parameters>Genetic Algorithm... 更改Maximum Number of evals为long。
Docking>Output>Lamarckian GA... 输入文件名,点击Save。
8. 运行AutoDock
Run>Run AutoDock... 点击Parameter Filename后的Browse,选择刚才生成的dpf文件,点击Open,点击Launch。
等待程序执行结束。
9. 分析结果
读取对接记录
Analyze>Docking>Open... 选择dlg文件,点击Open。
载入构象
Analyze>Conformations>Load...
按动画播放
Analyze>Conformations>Play,ranked by energy... 点击&,选中Show Info显示当前构象信息。
构象聚类
Analyze>Clusterings>Show... (默认Tolerances2.0)
重新聚类
Analyze>Clusterings>Recluster... Tolerances框中输入数值,可以多个数用空格隔开,再次执行上一步。
加入受体
Analyze>Macromolecule>Open... 选择pdbqt文件,点击Open。
各构象的中心用小球表示
Analyze>Docking>Show as Spheres...
显示相互作用
Analyze>Docking>Show Interactions
5.14 使用PISA进行生物大分子结构互作界面分析
简介(INTRODUCTION)
解析生物大分子结构以后,往往需要对结构中不同分子(链)之间的互作界面进行分析,得到分子间互作的关键信息,指导功能研究和应用。
本教程介绍了PDBePISA(简称PISA)在线服务器的使用,网址:http://pdbe.org/pisa/。这是一种基于网络的交互式工具,由PDBe提供,用于研究大分子复合物(蛋白质,DNA / RNA和配体)形成的稳定性。 除了对蛋白质数据库(PDB)中所有条目的表面、界面和组件进行详细分析外,该服务还允许上传和分析自己的PDB或mmCIF格式坐标文件。
生物系统中大分子复合物的稳定性基本上由以下物理化学性质决定:
1. 自由形成能
2. 溶剂化能量增益
3. 界面区域
4. 氢键
5. 界面上的盐桥
6. 疏水特异性
这些相互作用也可能在晶体系统中普遍存在,其中蛋白质分子在结晶溶液中彼此相互作用并将它们自身排列成有序的晶格以形成晶体。因此,考虑到一些结晶条件可以模拟溶液中的实际生物相互作用,晶体界面的分析和高阶结构的预测实际上可以反映生物系统中存在的那些。 PISA服务使用上面列出的所有标准来分析给定的结构并预测可能的稳定复杂性。但是,重要的是要强调PISA结果是预测,从中得出任何结论必须根据其他生物和实验证据。如果可以获得所研究蛋白质的结构,则PISA的结果可用于验证独立的生化数据。教程 (TUTORIAL)
1. 可以从PDBe页面上的多个位置访问PISA。您可以从PDBe主页(http://pdbe.org/)访问该服务,如下所示。
您还可以从特定条目的摘要页面访问特定条目的PISA。 单击左侧边栏上的链接转到PISA。 在出现的新页面上,选择“启动PDBePISA”。
这将打开提交表单。在提供的框中键入PDB Id code 1N2C。
一旦文件上传到服务器,它将为您提供有关PDB条目的初步信息(蛋白质链和结合配体的数量)。 条目1n2c具有8个蛋白质链和22个配体。最可能的组件称为8-mer。
2. 要了解有关此PDB条目的更多信息(例如蛋白质的名称、来源等),请转到此条目的摘要页面http://www.pdbe.org/1n2c/。
该条目的图册页面为我们提供了一个信息,即它是由ADP-四氟铝酸盐稳定的固氮酶复合物结构。 有三种不同的蛋白质NIFD(链A和B),NIFK(链C和D)和NIFH1(链E、F、G、H)。您可以单击蓝色的“查看”按钮(如上所示)来查看加载的PDB条目。
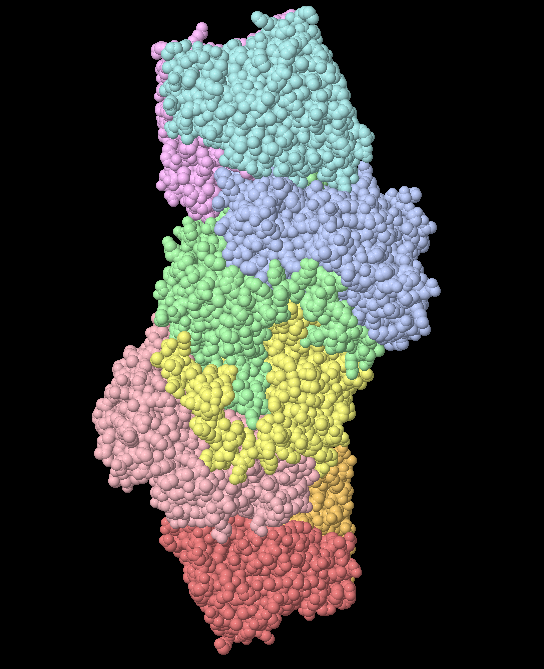
提交页面底部有三个绿色高亮显示的按钮:界面、单体和组件。它们中的每一个都提供与感兴趣的蛋白质相关的结构信息(结合能、溶剂化能、埋藏表面积、H键和盐桥等)。单体:
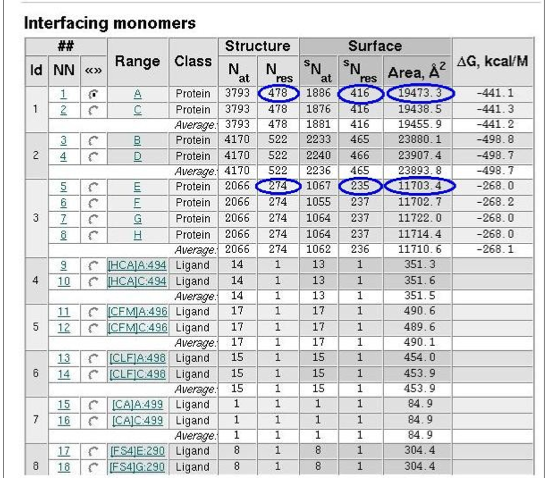
首先让我们从PDB条目中存在的不同单体开始。如果单击单体按钮,则会获得有关相应PDB条目的以下信息。
对于代表钼—铁蛋白的链A,蛋白质链中总共有478个氨基酸,其中416个是表面暴露的残基。该蛋白质19473.3Å2的溶剂可及区域和折叠的溶剂化能量(ΔG)为-441.1 Kcal / M.类似地,对于链E,总共有274个氨基酸,其中235个存在于蛋白质的表面上。该结构的溶剂可及表面积11703.4Å2和溶剂化能(ΔG)为-268 kcal / M.您还可以通过单击对应于蛋白质链的字母(A、B、C、D、E、F、G、H)在3-D图形查看器中查看单个蛋白质链。识别相互作用中的氨基酸残基: 单击结果页面上显示为数字的链接(突出显示上面的矩形)。在我们的例子中,链A为1,链E为5。 单击链A的链接A.这将带您进入下一页,您可以在其中获得溶剂可及性(solvent accessibility)信息。
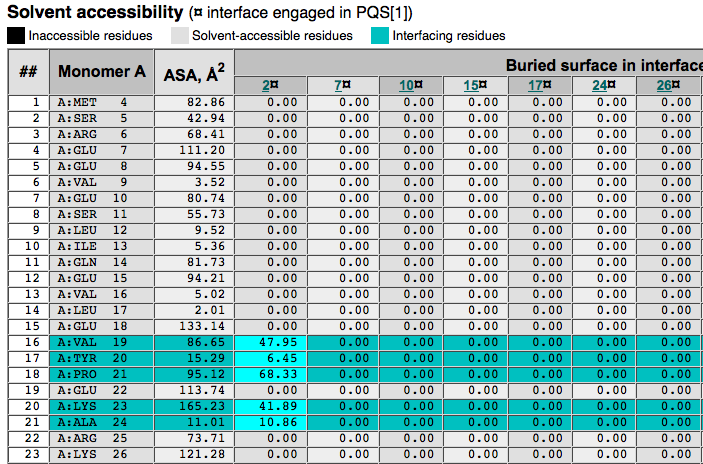
所有残基均根据其溶剂可及性进行颜色编码。 溶剂暴露的残基是灰色的,界面残基是蓝色的,埋藏的残基是黑色的。界面:现在让我们从页面顶部单击此PDB条目1n2c的界面按钮。

结果页面将为我们提供有关复杂结构中存在的两个蛋白质链之间的界面的详细信息。
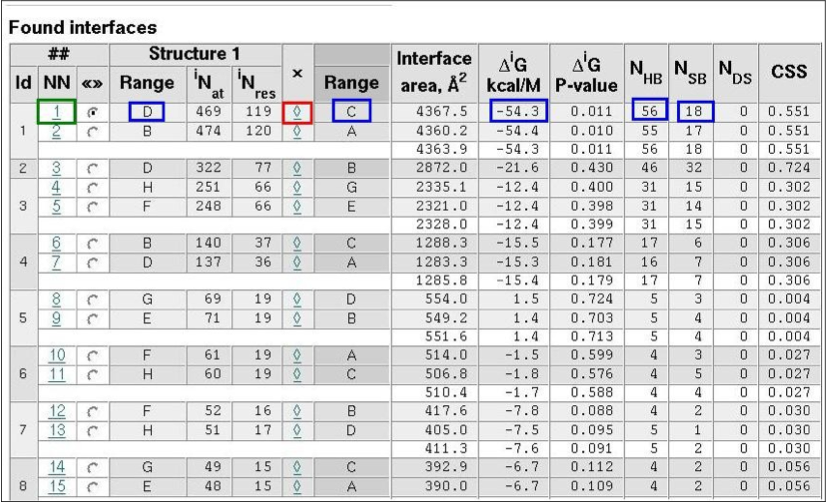
在上面的例子中,链C和D之间有18个盐桥和56个H-键相互作用。如果单击以红色突出显示的链接,则可以显示蛋白质链之间的界面区域。界面区域以红色和绿色突出显示,如下所示。

通过选择列名NN(以绿色突出显示)下的链接,可以找到关于复合物形成中涉及的特定残基的信息。
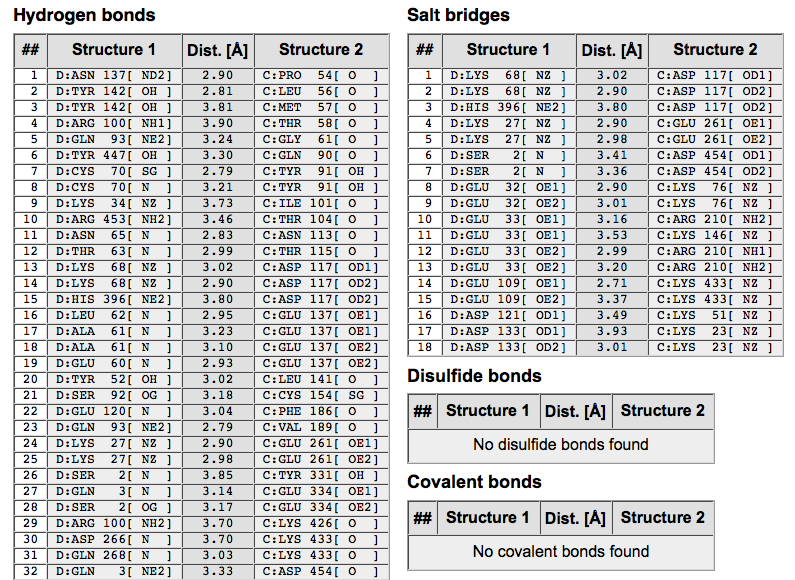
该页面还提供有关界面在复杂形成中的重要性的信息。
除了残基之间的盐桥和H键相互作用之外,结果页还提供了有关埋藏和可接触表面区域以及界面残基的溶剂化能量的信息。
该表中的所有残基均根据其溶剂可及性(灰色表示表面暴露、黑色表示埋藏、浅蓝色表示界面残基)进行颜色编码。 红色代表涉及氢/二硫键、盐桥或共价相互作用的残基。
组装模式:要获取1N2C的四元结构信息,请单击页面顶部的“程序集”按钮。
对于该条目,PISA提出的四元结构是异八聚体,其已经作为PDB文件中的稳定组装存在。 在装配结果页面中,PISA还提供有关埋藏和可接触表面区域的信息,以及在整个复杂结构形成时获得的溶剂化自由能。 单击上面显示的Id为蓝色以查看预测装配的详细信息。 单击第1组的“合成”部分以图形方式查看所形成的装配。
“View Dissociated”按钮将以图形方式显示组件的组合方式。在该条目的情况下,该程序表明链A、B、C、D形成组装的核心,然后链E、F、G和H结合在该核心蛋白的任一侧上以形成稳定的组装。
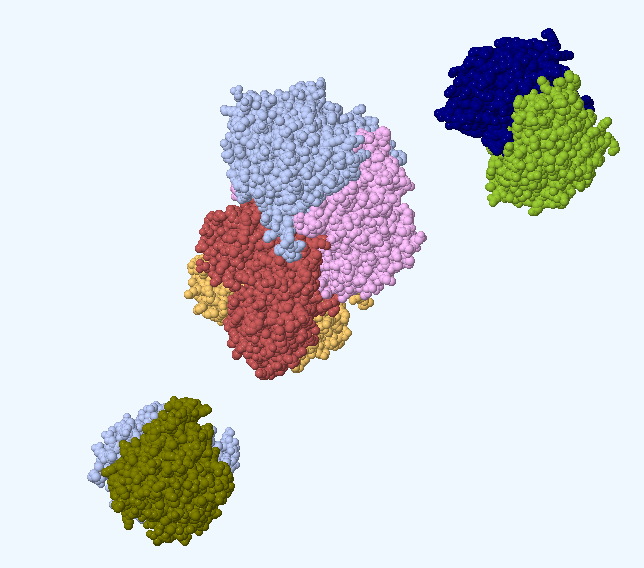
因此,使用PISA,您可以获得有关基于化学稳定性和晶体接触可形成的络合物类型的有价值信息。 PISA提供的残基信息可用于鉴定对形成可与生物学功能相关的稳定复合物至关重要的氨基酸。其他一些有趣的装配模式: 1DAT:提交的PDB是单链,但稳定的复合物是24聚体。


2WS9:PDB文件中的4个链。 实际装配240-mer。病毒在PDB中是特殊的,因为并非所有存在于所谓的晶体学不对称单元中的链都被提交。 相反,只有能够唯一描述二十面体重复单元的最小数量的链被提交在PDB中。 然后使用对称操作来产生完整的病毒衣壳。


3. PISA维护PDB中所有条目的数据库,可用于快速搜索符合特定标准的条目。这些可能是
(1)低聚状态
(2)对称/空间群
(3)盐桥和/或二硫键的数量
(4)UniProt / SCOP参考
(5)组成等
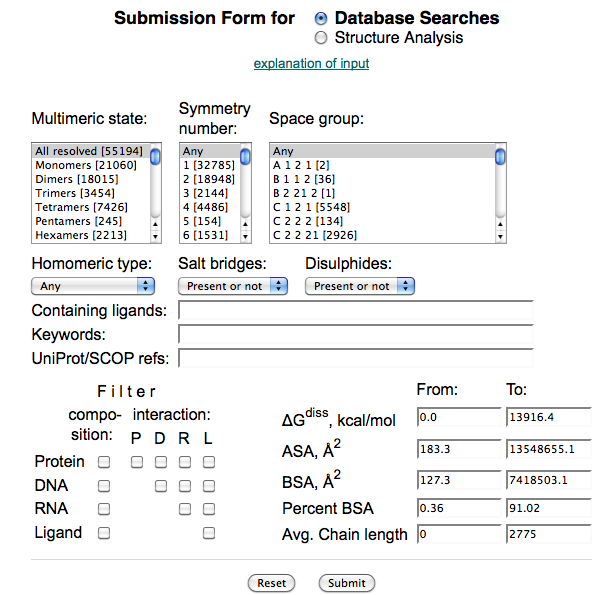
要查看数据库搜索表单,请从PISA提交表单页面中选择数据库搜索单选按钮,而不是结构分析按钮。为了搜索PDB中酶类“水解酶”并含有蛋白质和配体分子的所有条目,其中蛋白质与配体和蛋白质相互作用,并且预测的PISA组装是同型二聚体,提交形式可能看起来如下所示。
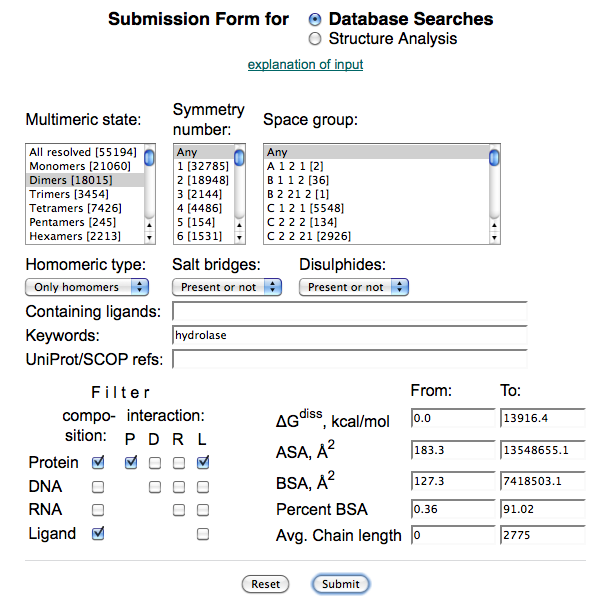
单击“提交”按钮以查看结果。
选择2rfp进行进一步分析。 单击第一列以查看界面的详细信息。单击mm Size列以图形方式查看,如下所示。
当您查看界面详细信息(如下所示)时,您还可以选择在整个PDB中搜索具有类似界面的其他结构。
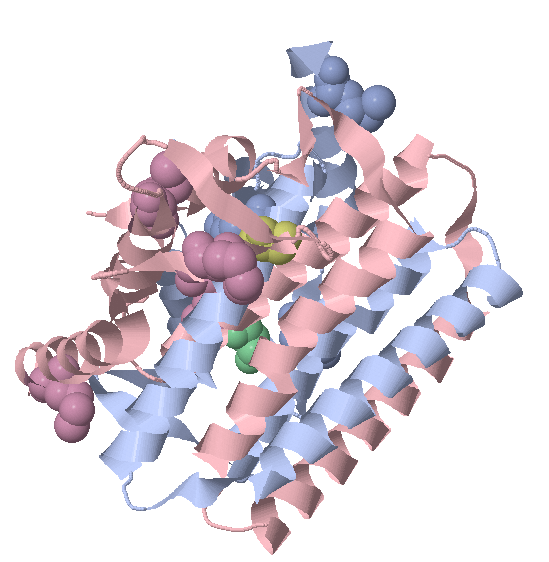

这是一次内存密集型搜索。选择此选项将显示另一个提交表单,其中可以调整某些参数以进行搜索。
搜索完成后,您将看到一个结果部分,其中包含与提交的条目具有类似界面的条目。
结果表明,有4个条目可能具有相似的界面(第一个与查询相同)。PDB条目2JIC是六角形,此条目的界面5可能与我们的查询匹配,但是Q分数非常低(0.018)。对于与相同界面无关的Q分数,范围从0到1。唯一具有高Q值的条目是相同蛋白质的条目。这表明在我们的查询结构中找到的此界面在给定的搜索条件下对于PDB是唯一的。更改查询参数可能会产生不同的结果。
5.15 Linux系统管理和常用命令
Linux的目录结构
特殊目录
根目录/
当前目录.
父目录..
Home目录~
路径名分绝对路径和相对路径。绝对路径名:从根目录开始的、以/分隔的各级目录名;
相对路径名:从当前目录开始的路径。
文件处理常用命令
1. man
即manual,查看某命令的帮助文件。
$ man ls
2. ls
即list,列出某一个目录下的所有内容。包括子目录和文件,按字母排序,不包括隐藏文件。如果后面没有跟任何路径,将显示出当前目录下的。
$ ls /home
一些参数:
-a:显示隐藏文件
-l:以列表形式显示更多信息
-t:按时间从新到旧排序
-r:倒序
$ ls -lrt /home
3. cp
即copy,复制文件或目录。
将文件f1复制为文件f2。
$ cp f1 f2
将文件f1复制到目录dir,文件名不变,可以同时复制多个文件。
$ cp f1 dir
$ cp f1 f2 f3 dir
一些参数:
-r:递归复制目录及各级子目录中所有内容。
$ cp -r dir1 dir2
4. mv
即move,移动或重命名文件。
将文件f1改名为f2。
$ mv f1 f2
将目录dir1移动到dir2。
$ mv dir1 dir2
将文件f1移动到目录dir,文件名不变,可以同时移动多个文件。。
$ mv f1 dir
$ mv f1 f2 f3 dir
一些参数:
-I 在移动时遇到同名文件均会询问。
-f 强制移动,同名文件直接覆盖。
5. rm
即remove,删除文件。
$ rm f1
而 rm 的参数比较常用的有几个: -f, -i, 与-r
-f:强制删除,不提出任何警告讯息。
-i:在删除每个文件之前均会询问。
-r:递归式删除目录及各级子目录中所有内容。
删除整个目录,小心不要随便使用
$ rm -rf dir
为防止误删除,推荐使用mv代替rm
6. mkdir
即make directory,创建一个目录。
$ mkdir dir
7. cd
即change directory,更改工作目录。
$ cd dir
$ cd ../..
8. rmdir
即remove directory,删除一个空目录。
$ rmdir dir
9. pwd
即print working directory,显示当前目录。
$ pwd
10. cat
即concatenate,查看文件内容。
$ cat file
11. more
将文件内容分页显示。按回车下行。按空格向后翻页。按q退出。按v编辑。
$ more file
12. chmod
即change mode,更改文件的权限模式。一个文件的权限分三组:文件的拥有者(u)、同群者(g)以及其他人(o)。每组权限有可读(r)可写(w)可执行(x)三种模式。每组满权项定义为rwx,缺的权项用-代替,三个组共需用9个字母表示。权限也可以用二进制表示:r—100; w—10; x—1,可把它们相加来表示不同组合,通常直接用一个八进制数字表示这三位二进制,如rwx—111—7,r-x—101—5,-wx—011—3。
设置rm的权限为755
$ chmod 755 /bin/rm
给file拥有者用户组加上rws权,其他用户删除ws权
$ chmod ug + rws, o – wx file
一些参数:
-R 递归所有子目录。
vi文本编辑器
打开文本文件
$ vi filename
vi编辑器支持编辑模式和命令模式,打开vi编辑器后自动进入命令模式。从编辑模式切换到命令模式使用“esc”键,从命令模式切换到编辑模式使用“A”、“a”、“O”、“o”、“I”、“i”键。
a:在当前字符后添加文本
A:在行末添加文本
i:在当前字符前插入文本
I:在行首插入文本
o:在当前行后面插入一空行
O:在当前行前面插入一空行
:wq保存并退出
:w保存
:q退出vi,要求没有任何修改,否则用下一条
:q!退出vi并放弃所有修改
/pattern从光标开始处向文件尾搜索pattern
?pattern从光标开始处向文件首搜索pattern
n在同一方向重复上一次搜索命令
N在反方向上重复上一次搜索命令
:s/p1/p2/g将当前行中所有p1均用p2替代
:n1,n2 s/p1/p2/g将第n1至n2行中所有p1均用p2替代
:g/p1/s//p2/g将文件中所有p1均用p2替换
:g/pattern/d将文件中所有含pattern的行删除
进程处理常用命令
1. ps
即process status,显示目前进程的状况。
显示所有进程
$ ps -A
显示指定用户的进程
$ ps -u root
显示所有进程信息,连同命令
$ ps -ef
与grep组合使用查找特定进程
$ ps -ef|grep ssh
2. top
动态显示系统当前的进程和系统处理器状况。将独占前台,直到按q键结束退出。可通过P(CPU使用时间)/M(内存使用量)/T(执行时间)键,选择不同行排序方式。
3. kill
杀死进程。
杀死PID为3434的进程
$ kill -9 3434
4. killall
按名字杀死进程。
$ killall xds
