4 细胞生物学实验技术
1.细胞间主要供本实验师生使用,其他需要使用细胞间的人员,需提前咨询金老师,联系相关负责人,以便进行安排。
2.所有使用细胞间的人员,均需要进行值日,一人/周。值日内容包括:检查桌面和清理垃圾、每周一次的大扫除(包括用消毒液拖地、清洗拖鞋、照射紫外30 min等)。细节见后面的描述。
3. 进出细胞间规则:
1) 进入细胞间前,在缓冲间更换专门的拖鞋或戴鞋套,进入细胞间后戴好手套,无菌操作时必须戴口罩,如有需要更换专门的实验服,切记不可将实验室的白大褂穿入细胞间。违反规定者,一次口头警告,一个月内被发现第二次,令其义务值日一周,情况恶劣的,一个月不得使用细胞间。
2) 进入细胞间实行严格的人物分流,人从缓冲间进入,物品从传递窗进入。
3) 禁止在细胞间内进食、饮水以及无事在细胞间内长时间逗留和交谈,违犯规定者,停用细胞间一个月。
4) 离开细胞间:首先确保带走所有实验垃圾(包括废液以及枪头盒报纸、离心管、EP管、手套等相关实验物品),移液枪调回最大刻度。同时谨记关闭显微镜、水浴锅、离心机电源,确保培养箱门以及展示柜的门关好,酒精擦拭超净台,关闭超净台通风和照明,并打开超净台紫外灯。
5) 爱护细胞间的仪器设备,按使用规则操作,不得随意拨动电器开关,显微镜用好后要登记,各功能部件复位;请节约使用实验材料,如不慎损坏了器材,应告知相应负责人登记;实验中发生意外事故时,应立即报告及时处理,切勿隐瞒或自作主张不按规定处理。
6) 对于培养基、PBS及血清:管理员负责管理配置培养基,所需人员网上填写领取申请,值日生负责分配,每人管理使用自己的培养基,禁止他人混用,造成污染。一经发现,直接取消细胞间使用资格一周。血清由管理员抽滤分装,放置于-20 °C冰箱,如若发现不多了,需及时告知管理者。
4.每天第一个使用细胞间者,需将超净台紫外灯打开照射15 min;每天最后一个离开细胞间的人,需确认洁净空调设备、超净台、离心机、水浴锅、显微镜等设备的电源已经关闭,检查冰箱、培养箱门是否关紧。并关闭照明,登记离开时间。每周的值日生负责打开细胞间大紫外灯,照射30 min,开灯前确定没有人和试剂(特别是培养基)暴露在紫外灯下。
培养箱使用规则
1.培养箱要定期进行灭菌,以便减少污染机会。将培养箱内的不锈钢板取出,包括4块横板和左右2块立着的架子,用酒精棉球清洁钢板和培养箱内壁;打开培养箱清洁内壁时动作要迅速,避免长时间敞开门使得大量不洁净空气进入,缩短过滤器的寿命;按照培养箱消毒程序进行消毒。
▲ 注:一般一个月需将培养箱消毒一次,可根据实验情况适当调整。
2.培养瓶皿放入培养箱前,用酒精消毒表面,并稍等至酒精挥发后再放入,以免培养箱内滞留过多乙醇蒸气。
3.从培养箱取放物品前,要用酒精清洁双手套,尽量缩短开门时间和减少开门次数。2人共用一层培养箱,按预定位置摆放培养器皿,方便查找,以免长时间敞开培养箱。
▲ 注:培养箱中的空气是经过过滤的洁净空气且保持恒温。长时间敞开或频繁开关培养箱,容易污染和降温,影响细胞状态。
4. 随时注意观察培养箱的CO2含量是否为5%,温度是否为37 °C,及时更换二氧化碳气瓶。发现培养箱出现异常,及时报告负责人并联系维修。
5. 未经他人允许,请勿移动和观看他人的细胞。
6. 如出现污染事件,及时向负责人报告,及时清理,擦拭培养箱。若出现严重污染,需清理培养箱内物品,对培养箱进行消毒处理。
▲ 注: 频繁将细胞拿出观察,容易给培养箱造成污染,同时也会影响细胞生长条件的恒定。
▲ 大瓶培养基预热建议使用水浴锅,50mL 离心管分装的培养基预热可喷洒酒精后置于培养箱中预热。
显微镜使用规则
1. 显微镜使用前,用酒精棉从中间至周围擦拭载物台。
2. 显微镜使用规定:打开电源——调节亮度旋钮——调节焦距旋钮,从低倍镜下看起,逐渐增大放大倍数。显微镜使用完毕后,放大倍数调到最小——焦距旋钮调到最低——亮度旋钮调到最小——关闭电源——使用登记。
3. 短时间内需再次使用显微镜时,为避免频繁开关显微镜电源,可不必关闭显微镜电源,但须将亮度旋钮调到最低;较长时间不需使用或离开细胞间时,须按规定关闭显微镜电源。
4. 明场使用无特别规定,需要特别提醒的是观察完毕立即关闭电源。为了延长汞灯使用寿命,荧光观察的同学需要严格执行登记制度,即使用日期、人员姓名、使用前以及使用完毕后的汞灯读数,使用的起始时间以及结束时间,为了延长汞灯的寿命,应避免频繁开关,每次汞灯开机至少半小时才能关闭,关掉半小时以上才能再次打开。
5. 请严格按规定使用显微镜,以增加显微镜的寿命。违反规定者,停止使用细胞室一个月。
超净台相关操作规则
1. 超净台的使用暂时不需要预约。
2. 超净台每天第一个使用者可以用紫外灯照射15分钟灭菌,其后使用后用紫外灯照射15分钟灭菌,以便下一位使用者直接使用。使用前、后需用酒精棉球擦拭工作区消毒,所有物品放入超净台内使用前均应消毒。
3. 75%酒精由值日生负责补给,发现细胞间内酒精灯酒精存放量不足时请及时通知值日生。
4. 垃圾分类:超净台中应摆放废液桶一只,用于暂时存放废弃物,使用者使用时需要套上保鲜袋或EP手套,实验结束后将袋内垃圾倒入黑色垃圾袋中,确保废液杯呈干净状态,若发生漏液需将废液杯冲洗晾干备用,细胞房内的垃圾袋快满时,最后一个人将垃圾带出细胞间。另外细胞间的垃圾桶只能扔酒精棉,口罩和手套,严禁倒入液体和使用过的枪头(这些垃圾是统一倒入黑色垃圾袋中,随时并及时带出细胞间)。使用者实验结束后若发现垃圾桶已装满,请通知值日生或顺手将桶里的垃圾袋带出细胞房。
5. 超净台物品摆放整齐,用后回归原位。
值日生工作职责
值日生需要认真完成以下工作,以便接收包括管理员在内的所有使用人员的监督,如果有两人及以上使用人员向管理员提出抱怨,则该值日生继续值日一周,以此类推。
1. 每周值日生时间从周二开始,至下周一结束。结束值日前需保证:
2. 清洗水浴锅,换水。水浴锅的清洗:水浴锅内胆用酒精喷洒,擦净,换水烧沸至100 °C后,保持1 h后将水浴锅调回37 °C,切记。
3. 培养箱:准备两瓶灭菌的双蒸水。待冷却后,将培养箱最下层的水箱里的水换成灭菌双蒸水,并用酒精擦拭培养箱内部。
4. 酒精:给细胞房内添加足够的75%的酒精于15 L桶中。(此项按照已排好的值日表两人一组完成,并非每周需要配置)
▲ 注:擦手消毒用75%的酒精,可以用瓶装95%酒精或瓶装无水乙醇配制;擦拭台面消毒用75%的酒精可以用工业酒精配置;酒精灯直接用95%的工业酒精即可,向喷壶或灯内添加酒精时请留意。)
5. PBS:配置一瓶PBS,灭菌后置于细胞间展示柜中,待分装。
6. 枪头:务必保证工作台上非无菌的枪头的供应,即将工作台上的空枪头盒补满枪头。将用完的枪头盒取回放于实验室固定的地点。
7. 值日周内每天都需要:开大紫外灯消毒细胞房15-30 min;更换垃圾袋;检查桌面和超净台面是否收拾干净;检查培养箱内水量是否足够。
8.(注:若上周值日生及时给培养箱内水槽加足水,根据经验未来一周内培养箱不会干水,但值日生仍应经常检查,以便及时发现异常情况。)
9. 摇床:缓冲间的摇床排水需要及时倾倒。
10. 泵:及时倾倒泵中液体,并于瓶底加入适量洁尔灭。
11. 清洁:及时打扫细胞房。包括拖地,擦桌子、清洁和整理抽屉,擦超净台、冰箱及桌上的仪器,给水浴锅加水或换水,洗细胞房专用的实验服和拖鞋,紫外消毒细胞房。
其他注意事项(使用细胞间人员必须遵守):
1. 禁止浪费培养基,禁止长时间(超过一天)将一整瓶培养基放置在室内。
2. 使用过的物品归位,例如超净台的EP管和枪头盒都要封口以免污染,移液器量程归位并放置整齐,电移液器及时充电。
3. 不随意将手套,锡箔纸以及其他垃圾扔在地面,桌面以及其他仪器上方。
4. 使用过后的瓶子,管子,枪头盒随身带走,不要遗留在细胞间。
5. 禁止过多过早地积囤细胞间耗材(如注射器,滤膜等)或试剂,发现耗材或试剂快被用完时请及时登记购买,每个人都应该有责任感。
4.2 细胞系的冻存与复苏
简介(INTRODUCTION)
细胞系的冻存是将生长较好的传代细胞悬浮在加有冷冻保护剂的溶液中,以一定的冷冻速率降至零下某一温度,并在此温度下对其长期保存的过程;而复苏就是以一定的复温速率将冻存的细胞系恢复到常温的过程。我们的实验原则是“慢冻快融”,这样可最大限度的保存细胞活力。如果冷冻过程得当,一般生物样品在液氮中可保存十年以上,在-80 °C冰箱可保存半年左右。
材料(MATERIALS)
r 试剂(REAGENTS)
DMSO(二甲基亚砜)、小牛血清、液体培养基、1×PBS、trypsin胰蛋白酶消化液
r 试剂准备(REAGENT SETUP)
1. 培养基/小牛血清/DMSO为5:4:1;以配制 1 mL冻存液为例,加入500 μL新鲜培养基,400 μL小牛血清,100 μL DMSO,最好现做现配,也可配好后放在4 °C保存一周,对于一些培养条件苛刻的细胞可以适当加大小牛血清的比例。
2. 10% DMSO冻存液:以配制1 mL为例,900 μL培养基,加入100 μL纯DMSO,悬浮混匀。
3. 10% DMSO血清冻存液:以配制1 mL为例,900 μL FBS小牛血清,加入100 μL纯DMSO,悬浮混匀。
4. 1×PBS:NaCl 8 g,KCl 0.2 g,Na2HPO4·12H2O 3.63 g,KH2PO4 0.24 g,溶于900 mL超纯水中,用盐酸调pH值至7.4,加水定容至1 L,高温灭菌后常温保存。
5. 消化液:0.25 g的胰蛋白酶和0.02 g的EDTA加入1×PBS,终体积为100 mL,pH 7.4,0.22 μm滤过除菌分装,4 °C保存(目前本实验室购买现成trypsin分装使用,该胰蛋白酶浓度较高,建议用裸培养基稀释10倍再用)。
r 器械(EQUIPMENT)
冻存管、离心机、细胞培养箱、4 °C冰箱、-20 °C冰箱、-80 °C冰箱、冻存盒、液氮冻存罐
实验步骤(PROCEDURE)
1. 细胞冻存 ►预期时间消耗:30 min
2) 从细胞培养箱中拿出处于对数生长期的细胞(10 cm大皿长至80~90%),弃去上清加入生理盐水洗涤,弃上清并重复洗涤一遍。
3) 加入1 mL胰酶消化液,将大皿放于培养箱中孵育1 min,待细胞呈现大片掉落时,加入6 mL完全培养基,迅速吹打培养皿底,将细胞移入离心管中,130 g离心2 min。
4) 弃上清,用冻存液吹打细胞(长满的大皿需要5~10 mL冻存液,使浓度达到3~6×106/mL)再移至冻存管中(一个冻存管1 mL),做好标记,包括细胞名称,冻存日期及姓名。
5) 冻存管放置冻存盒中,将冻存盒放入-20 °C冰箱2 h,然后放入-80 °C冰箱中过夜,次日取出冻存管,移入液氮容器内。
▲ 消化时间不易过长,最长时间不超过五分钟,不同细胞类型所需时间不一样,比如293T贴壁不牢,不需要用胰酶消化,但是像Hela细胞则需要用胰酶消化。
2. 细胞复苏 ►预期时间消耗:30 min
1) 提前准备37 °C millipore水。
2) 从液氮中取出冻存管,迅速放在37 °C水浴至融化,期间需要不断摆动助融。
3) 130 g离心2 min离心弃上清,用1 mL完全培养基轻轻吹打并移至60 mm培养皿,再补加完全培养基至5 mL,混合均匀后放入培养箱。
4) 次日,换成新鲜的培养液。
▲ 从液氮拿出的细胞需要迅速放于37 °C 水浴,并不断晃动。
针对性建议(TROUBLESHOOTING)
1. 冻存时细胞状态需要良好,否则复苏时存活率低;一般使用10% DMSO全血清的冻存液,细胞状态较好,虽然有点浪费血清。
2. 冻存时讲究慢冻,小伙伴们将冻存盒放冰箱的时间控制好,不要忘了。
3. 冻存时细胞浓度控制在3~6×106/mL;太多或太少的细胞量都不利于细胞的存活。
4.3 细胞系的传代与培养
简介(INTRODUCTION)
细胞在培养瓶长成致密单层后,已基本上饱和,为使细胞能继续生长,同时也将细胞数量扩大,须进行传代再培养。传代培养也是一种将细胞种保存下去的方法。同时也是利用培养细胞进行各种实验的必经过程。悬浮型细胞直接分瓶就可以,而贴壁细胞需经消化后才能分瓶。
材料(MATERIALS)
r 试剂(REAGENTS)
培养基(RPMI 1640、DMEM、SIM SF)、胎牛血清、双抗青链霉素、胰蛋白酶消化液、生理盐水
r 实验前准备(SETUP)
完全培养基:90%液体培养基+10%胎牛血清+1%双抗青链霉素
胰蛋白酶消化液:本实验室目前使用直接购买的trypsin分装冻存
r 器械(EQUIPMENT)
EP管、15 mL离心管、移液枪、离心机、细胞培养箱
实验步骤(PROCEDURE)
► 贴壁细胞传代与培养 ►预计消耗时间:30 min
1. 倒置显微镜下观察细胞形态,确定细胞是否需要传代,并将培养基置于37 °C培养箱预热;胰酶消化液室温放置。
2. 用真空泵抽去培养皿中上清液,加入生理盐水洗涤两遍,除去悬浮的死细胞以及去除培养基,然后加入胰酶消化液1 mL,轻轻摇晃后放于细胞培养箱中1 min(可酌情增加时长,随时肉眼观察细胞的脱落情况),也可以放在倒置显微镜下观察,当细胞收回突起变圆即可,或晃动培养皿细胞呈现大片脱落时即可;
3. 加入2 mL培养基并迅速吹打培养皿底,将细胞吹散均匀后移入离心管中,用完全培养基补加至10 mL,130 g离心2 min。
4. 弃上清,加入1 mL完全培养基吹打均匀,取适量的细胞数加入到新的培养基中,摇晃均匀。
5. 放置培养箱中培养,可以每天观察,期间如果细胞培养基营养不够或者变黄时可更换成新鲜培养基。
▲ 关键步骤:
♫ 控制好消化时间,有些贴壁细胞贴壁较紧,需要确保吹打均匀,否则细胞会聚团,不能呈现单个细胞贴壁,影响实验。重悬后的细胞可以取1/5放入新的培养基中,这可以根据实际情况操作,如果细胞多可以少取一些,细胞少就多取一些。
► 悬浮细胞传代与培养 ►预计消耗时间:30 min
1. 倒置显微镜下观察细胞形态,确定细胞是否需要传代,并将完全培养基置于37 °C培养箱。
2. 将培养瓶中的细胞用移液管转移至15 mL离心管中,130 g离心2 min。
3. 弃上清,加入1 mL完全培养基,取适量细胞加入到新的培养基中,摇晃均匀。
▲ 关键步骤:
♫ 吹打细胞需要轻柔,并且混合均匀。重悬后的细胞可以取1/5放入新的培养基中,这可以根据实际情况操作,如果细胞多可以少取一些,细胞少就多取一些。
针对性建议(TROUBLESHOOTING)
本实验室常用于蛋白表达的有哺乳动物细胞HEK 293T和昆虫细胞Sf9、Hi5,其中:
HEK 293T细胞使用90%DMEM+10%胎牛血清+1%双抗青链霉素,是贴壁细胞,可以直接从培养皿底部吹打下来,所以传代时不需要使用胰酶消化。Sf9、Hi5细胞使用SIM SF+1%双抗青链霉素,是悬浮细胞,传代时直接分瓶,每次传代前都需要计数,细胞密度大概在2×106 g/mL时可以传代,传代后细胞密度为0.5×106 g/mL。
细胞培养条件会根据不同细胞要求不同,以下是总结的条件:
1. HEK 293T:人肾上皮细胞;10% FBS DMEM培养,可以直接从培养皿板底吹下来,不需要用胰酶消化。
2. BHK‐21:幼地鼠肾细胞;10% FBS DMEM培养,胰酶消化传代。
3. NK92:人的 NK 细胞系(悬浮细胞系);12.5% FBS +12.5% horse serum α‐MEM,100U/mL 的 IL‐2。
4. MCF‐7:人乳腺癌细胞;10% FBS DMEM培养,用生理盐水洗两次胰酶消化传代。
5. L-929:小鼠成纤维细胞;1:3 传代,扩大培养取上清。10% FBS 1640培养基培养,长满后取走上清,换新鲜培养基,过 2~3 天可以取用,一共可以取两批。
6. HT‐29:结肠癌细胞;10% FBS DMEM培养,生理盐水冲洗两次胰酶消化传代。
7. DC2.4:小鼠树突状细胞;10% FBS DMEM培养,胰酶消化传代。
8. HeLa:人宫颈癌细胞;10% FBS DMEM培养,生理盐水冲洗两次并胰酶消化传代。取1/5总量长三天就能长满大皿(10 cm dish)。
9. THP-1:人单核细胞系,10% FBS 1640培养基培养,3天一代,细胞长过影响状态,小瓶留 2 mL 传代,大瓶留 7 mL传代。
10. HepG2:人肝癌细胞系;10% FBS DMEM培养,生理盐水冲洗两次胰酶消化传代。
11. L-O2:人正常肝细胞;10% FBS DMEM培养,生理盐水冲洗两次胰酶消化传代。
12. Huh 7:人肝癌细胞系;10% FBS DMEM培养,生理盐水冲洗两次胰酶消化传代。
13. Hepa1-6:小鼠肝癌细胞系,10% FBS DMEM培养,生理盐水冲洗两次胰酶消化传代。
14. NCTC 1149:小鼠肝脏细胞系,10% horse serum DMEM培养,生理盐水冲洗两次胰酶消化传代。
4.4 细胞系转染
简介(INTRODUCTION)
转染是将外源性基因导入细胞内的一种专门技术。转染大致可分为物理介导、化学介导和生物介导三类途径。电穿孔法、显微注射和基因枪属于通过物理方法将基因导入细胞的范例;化学介导方法中脂质体转染方法较为常用;生物介导方法现在比较多见的各种病毒介导的转染技术。理想细胞转染方法,应该具有转染效率高、细胞毒性小等优点。病毒介导的转染技术,是目前转染效率最高的方法,同时具有细胞毒性很低的优势。但是,病毒转染方法的准备程序复杂,在敲低细胞系构建时详述。这章以介绍脂质体转染为主。脂质体是利用脂质膜包裹DNA,借助脂质膜将DNA导入细胞膜内。带正电的阳离子脂质体,DNA并没有预先包埋在脂质体中,而是带负电的DNA自动结合到带正电的脂质体上,形成DNA-阳离子脂质体复合物,从而吸附到带负电的细胞膜表面,经过内吞被导入细胞。
材料(MATERIALS)
r 试剂(REAGENTS)
Lipo 6000(Beyotime)、完全培养基、Opti-MEM培养基(裸培养基)、不含双抗的完全培养基、目的质粒
r 实验前准备(SETUP)
去内毒素质粒小抽试剂盒
r 器械(EQUIPMENT)
Ep管,离心机,CO2细胞培养箱,细胞培养板,Timer
实验步骤(PROCEDURE)
► 细胞转染 ►预计消耗时间:2 Day
1. 根据实验要求选择合适的细胞培养板,在这里我们以6孔板为例,其他培养板体积与使用DNA量见下表:
不同体积细胞中试剂和DNA推荐使用量
| 96-well | 48-well | 24-well | 12-well | 6-well | 6 cm dish | 10 cm dish |
Lipo6000 | 0.2 μL | 0.5 μL | 1 μL | 2 μL | 5 μL | 10 μL | 30 μL |
裸培养基 | 5 μL | 12.5 μL | 25 μL | 50 μL | 125 μL | 250 μL | 750 μL |
DNA | 100 ng | 250 ng | 500 ng | 1 μg | 2.5 μg | 5 μg | 15 μg |
裸培养基 | 5 μL | 12.5 μL | 25 μL | 50 μL | 125 μL | 250 μL | 750 μL |
稀释好的Lipo6000和DNA分别在室温静置5分钟,随后混匀并室温静置20分钟 | |||||||
每孔加入的 混合物量 | 10 μL | 25 μL | 50 μL | 100 μL | 250 μL | 500 μL | 1500 μL |
4-6小时候更换培养液 | |||||||
2. 转染前一天将传代的细胞传到细胞培养板每个孔中,使每孔的细胞数达到60~70%。
3. 转染之前1~2 h更换无双抗已预热的完全培养基,继续放入培养箱中培养。
4. 准备好两个无菌的Ep管,将lipo6000和质粒分别用裸培养基稀释,室温放置5分钟,混匀再室温放置20分钟。注意不要涡旋混匀。
5. 将混匀好的混合物加到孔板中的细胞悬液中,培养4~6小时,更换成完全培养基。
6. 转染后24~48小时后可以检测各项指标,如通过WB、ELISA、流式细胞数、IF等等。
▲ 关键步骤:
♫ 细胞分板时控制好细胞密度,这需要根据不同细胞的特性,最好根据经验总结一下最适细胞密度。
♫ 如果需要爬片,建议不要用太高的细胞密度,否则在固定时成片地脱片。
♫ 如果转染后的细胞状态极差,适当改变lipo和质粒的量,过多的lipo或者质粒均对细胞有毒性。
♫ 转染时可以增加一组阳性对照,转入表达荧光蛋白的质粒,根据细胞是否发荧光来判断转染效果。
针对性建议(TROUBLESHOOTING)
1. 如果转染后细胞状态很差,转染后更换培养基时可以用5%双抗的培养基来替代完全培养基。
2. 不同的细胞系转染效率都不同,需要自行摸索浓度和时间。
3. 对于NIH3T3、CHO、HEK293T等细胞系,推荐在转染后6小时更换培养液。
4. 本实验使用碧云天的lipo6000,转染时不受培养液中的血清和抗生素的影响,因此使用不含双抗但含有血清的细胞培养基;若是使用Invitrogen公司的Lipo2000转染试剂,对血清比较敏感,因此需要使用不含双抗生素,不含血清的培养基进行转染,或直接用OPTI-MEM。
4.5 用PEI于贴壁细胞瞬转
简介(INTRODUCTION)
真核细胞表达具有先天的优势,但是大规模质粒转染,存在成本、效率、毒性等挑战,一定程度上,阻碍了真核表达体系用于大规模蛋白表达。其中一种有效的方案就是采用Polyethylenimine (PEI),又名聚乙烯亚胺,作为转染试剂,具有多方面的优势。由于PEI这种阳离子聚合物,能够与带有负电的DNA分子结合,形成大小合适的复合物,适合与细胞膜相互作用,进入细胞。
材料(MATERIALS)
r 试剂(REAGENTS)
细胞系、培养基、血清、抗生素、PEI粉末(high molecular weight, MW 25 kDa, water free (Aldrich, cat #408727)or Polysciences (cat# 23966-2))
实验步骤(PROCEDURE)
1. Preparation of PEI solution (1 mg/mL, pH 7.0): dissolve PEI powder in appropriate amount of distilled water (heat water to 90C could facilitate dissolving PEI). After solution cools down to room temp, adjust pH to 7.0 by HCl. Filter sterilize (0.22 μm), aliquot and store at -20°C or -80°C; a working stock can be kept at 4°C.
2. 293EBNA (or C18) cell line is maintained in IMDM (has biotin and Vitamin B12) or DMEM medium with 10% FBS, 1x Non-essential amino acid, 1% Pen/strep plus Geneticin/G-418.
3. 24 hour before transfection, seed 3x106 293EBNA cells in 150mm cell culture grade Petri dish or 175 cm2 T-flask with 20 mL warm medium. One can scale this based on the size of the flasks used.
4. Just before transfection, take 2 mL IMDM/DMEM medium (without any SERUM!!!), warm up and add in 16 μg plasmid DNA from Qiagen Endo-free kit. (The DNA quality is very important: the OD260/OD280 should always exceed 1.8). Mix briefly.
1) Directly add 64 μL PEI solution (1 mg/mL) into DNA/medium. Vortex for 10 sec. (Maintain DNA/PEI 1:4, this is most optimal ratio I tested; one should also test different amount of PEI to find the optimal conditions for a particular vector, i.e., a vector with or without oriP may require different optimized amount of PEI and DNA)
2) Incubate DNA/PEI mix at room temperature for 15 min.
3) Add DNA/PEI mix in one 293EBNA plate drop by drop. Then shake well to mix the medium.
5. Culture cells for 72 hrs.
6. Harvest cells and wash them in PBS three times to remove serum or media components or trypsin/EDTA that may inhibit metal-affinity chromatography. Or, one can transfer cells in antibiotic containing media to select for populations of transfected cells, which can take an additional week.
▲ 关键步骤:
♫ Scale up accordingly for more plates. (I do 10 plates in 20 mL IMDM in 50 mL conical tube, but no more than 20 mL in one tube).
♫ Transfection efficiency can be 50~80%. FACS peaks for transfected cells are sharper than calcium phosphate transfected cells.
♫ Transfection cocktail should NOT contain serum. Cells grown in serum-containing media can be transfected by adding the transfection cocktail (NO SERUM) to cells.
♫ The optimal amount of PEI and DNA should be determined by considering the morphology of the cells (toxicity), the number of antibiotic-resistant clonies appeared, the total amount of proteins expressed, etc.
♫ Generally, non-oriP containing plasmids require more DNA to achieve the best expression compared with oriP containing plasmids, such as pCEP4, in HEK293 C18 (EBNA) cells.
4.6 细胞系基因敲低技术
目前实验室常用的基因敲低技术有三种,分别是shRNA,siRNA和CRISPR/Cas 系统;shRNA和siRNA都是在RNA水平上降低蛋白的表达,CRISPR/Cas是在DNA水平上降低蛋白的表达;shRNA与siRNA的加工方式相同,在细胞核表达后被Drosha加工然后由Exportin-5蛋白转运到细胞质中,在细胞质中它们与Dicer结合去除环状序列。在与RISC结合并去掉其中一条RNA链后,它们识别mRNA占有互补序列,导致mRNA降解。CRISPR/Cas系统是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御,它可以将入侵的噬菌体和外源质粒DNA片段整合到规律成簇的间隔短回文重复序列crisper中,随后被内切酶切割形成crRNA,通过碱基互补配对形成PAM互补区,指导cas9内切酶来对DNA进行定点切割,从而达到在DNA水平上降低蛋白表达的目的。
材料(MATERIALS)
r 试剂(REAGENTS)
慢病毒包装质粒、LB、氨苄青霉素、Amp板子、小抽试剂盒、完全培养基、减血清培养基(Opti‐MEM)、lipo2000、嘌呤霉素、细胞培养皿(10 cm)、24孔板、12孔板、6 孔板、9孔板、0.45 μm滤膜
r 试剂准备(REAGENT SETUP)
LB:已经灭菌的新鲜LB(无抗);
氨苄青霉素(1000×):用ddH2O配制成终浓度为100 mg/mL,用0.22 μm滤膜过滤后分装;
嘌呤霉素:根据需要的浓度自行配制。
r 器械(EQUIPMENT)
EP管、冻存管、离心机、细胞培养箱、-80 °C冰箱
实验步骤(PROCEDURE)
► shRNA敲低细胞系
1. 细胞嘌呤霉素浓度确定实验:
1) 24孔板内以5~8×104 cells/孔的密度铺板,铺足够量的孔以进行后续的梯度实验。细胞孵育过夜。
2) 准备筛选培养基-含不同浓度嘌呤霉素的新鲜培养基(如0~15 μg/mL,至少5个梯度)。
3) 细胞孵育过夜后加入筛选培养基,孵育细胞。
4) 约2~3天更换新鲜的筛选培养基。
5) 每日监测细胞观察存活细胞比例。嘌呤霉素的最佳作用时间一般在1~4天之间。
6) 最小的抗生素使用浓度就是指从抗生素筛选开始1~4天内杀死所用细胞的最低筛选浓度。
2. 在我院shRNA库中查找有没有所需基因的shRNA,一般会有4~5条,将shRNA信息复制另存到excel表格中,发送到邮箱wumianlab@163.com。
3. 拿到shRNA后加入LB摇菌小抽后继续转化后中抽,得到浓度较高的质粒。
4. 第一天晚上接种 293T 于6孔板,每孔接种70~80%(1×106个),并加入2 mL培养基,每种 shRNA对应一孔。
5. 第二天早上转染质粒:2 μg shRNA, vsv-g 1 μg, gag 2 μg, Rev 2 μg,lipo2000 18 μL。
6. 第三天早上弃上清,加入1 mL完全培养基。
7. 第四天吸出含病毒的培养基上清液,4000 rpm/min 离心 5 min,吸上清用0.45 μm滤膜过滤后放入无菌的EP管中,-80 °C保存(可保存六个月),也可马上使用。
8. 第三天的时候可同时将目的细胞接12孔板,密度大约为5×105/mL。
9. 第四天将12孔板中培养基上清去除,加入病毒上清1 mL,感染6 h后补加1 mL新鲜培养基。
10. 第五天更换筛选培养基,第七天换液(利用含有嘌呤霉素的培养基筛选)等细胞长满后将细胞移至6孔板,继续加入嘌呤霉素维持,此为瞬敲细胞系。
11. 稳敲细胞系(筛选时嘌呤霉素浓度不变)是将瞬敲得到的细胞稀释成10 cells/mL,转移至96孔板中培养,每孔100 μL(大约每孔1个细胞)。
12. 18~24 h后,观察每孔的细胞数,将只有一个细胞的孔做上标记。
13. 观察做标记的孔形成的克隆数,只形成1个克隆的被认为是单克隆。
14. 持续观察确认孔中克隆数,当96孔板中的单克隆达到较高密度时,转移至12孔板培养。
15. 细胞在12孔板中达到较高密度时,转移至6孔板中继续培养,同时取少量细胞鉴定效果(western blot 或QPCR)。
16. 确认表达后,将单克隆转移至10 cm培养皿中培养,一般需要2-4周细胞才能达到较高密度。
17. 冻存细胞(冻存液不含抗生素)。
18. 稳转细胞系建立以后,可降低抗生素浓度(如降成原来的一半)。
▲ 不同的细胞种类对于接板时细胞数以及抗生素的浓度的选择不同,需要自己摸索或者请教师兄师姐。
► siRNA敲低细胞系
(以下步骤所有的量和浓度都是以孔为基础)
1. 转染前一天,接种适当数量的细胞至细胞培养板中,使转染时的细胞密度能够达到 30~50% (不同细胞生长速度不一样,因此,接种细胞的数量需要根据细胞培养的经验),请使用无抗生素的培养基。
2. 对于每个转染样品,按照如下操作准备lipo2000‐siRNA混合物:
1) 稀释转染试剂 lipo2000:使用前,将lipo2000转染试剂轻轻摇匀,然后取 2 μL,用100 μL不含血清的Opti‐MEM稀释,轻轻混和,室温孵育5 min;。
2) 稀释 siRNA:用100 μL Opti‐MEM稀释40 pmol的siRNA,轻轻混和。(对于 stock 浓度为 20 μM 的 siRNA,加2 μL即可)
3) 稀释好的lipo2000经过5 min的孵育后,与上述(b)稀释好的siRNA轻轻混和,室温培养20 min以形成siRNA‐lipo2000混和物,溶液可能会有浑浊,不过不会影响转染。
4) 将siRNA‐lipo2000混合液加入含有细胞以及培养液的细胞培养板中,轻轻摇晃,使之混和。
3. 将培养板置于37 °C的CO2培养箱中培养至检测时间(24~96 h)。沉默效率的检测一般建议的时间为24~72 h。
▲ 关键步骤:转染操作完成,经过37 °C培养 4~6 h后,可以将孔里含有siRNA‐lipo2000混合液的培养基移去,更换新鲜培养基,这样也不会影响转染的效率。注意:如果转染时使用的是不含血清的培养基(即血清饥饿的条件下进行转染),4~6 h后必须换成完全培养基(含血清、含抗生素),以确保细胞生长。
► CRISPR/Cas9敲低细胞系
预期时间消耗:两个星期间断性实验
1. 通过软件设计基因的sgRNA并送到公司合成。
2. 拿到的sgRNA(上游,下游)需要复性,体系:sgRNA(F 100 μM)2 μL,sgRNA(R 100 μM)2 μL,NEB buffer2(sigma)2 μL,ddH2O 14 μL,95 °C放置5 min,然后自然冷却至室温。
3. cas9载体质粒酶切,体系:cas9载体质粒5 μg,bsmb1内切酶3 μL,FastAP 3 μL,buffer 6 μL,ddH2O补齐至60 μL在37 °C放置30 min,然后0.8%核酸胶跑胶,可以看到两条带,分子量大的那条胶回收。
4. cas9质粒酶切后回收产物和复性的sgRNA连接,使用T4连接酶,用量和步骤见T4连接酶说明书,连接产物转化,挑点摇菌,鉴定后测序抽质粒。
5. 得到的质粒与shRNA敲低方式相同。
▲关键步骤:酶切需要彻底,并且需要设计出具有敲除效果的sgRNA。
针对性建议(TROUBLESHOOTING)
1. 细胞系敲低时细胞状态不稳定会在感染时大面积死亡,所以无论是293T还是目的细胞状态都需要非常良好。
2. 对于抗生素浓度以及病毒滴度最好能事先做一下预实验确定。
3. 重复检测具有敲低效果的细胞系需要马上冻存保种避免重新筛选,这一点很重要。
4. 慢病毒包装得到的病毒最好现用,放于-80°C冰箱的病毒上清应避免反复冻融。
4.7 NF-κB启动子活性检测
简介(INTRODUCTION)
转录因子可以结合目的序列的启动子区域从而激活下游分子的转录。目前市场上已有很多种商品化的启动子活性检测相关的质粒,其中本实验室常用的为NF-κB启动子。pNF-κB质粒中已包含了五个拷贝的其启动子序列,而下游为萤火虫萤光素酶(Firefly luciferase)基因,因此一旦NF-κB被启动,即可以在细胞中表达萤火虫萤光素酶。与pNF-κB同时转染的还有pRenilla质粒,它可以持续性表达海肾萤光素酶(Renilla luciferase),因此可作为内参。
我们可以使用双萤光素报告基因检测试剂盒,分别检测萤火虫荧光素酶和海肾萤光素酶的表达量。先以萤光素(luciferin)为底物检测萤火虫萤光素酶,后以肠腔素(coelenterazine)来检测海肾萤光素酶,并且在后续加入海肾萤光素酶底物时,同时加入抑制萤火虫萤光素催化luciferin发光的物质,使后续检测仅仅检测到海肾萤光素酶的活性,实现双萤光素酶报告基因的检测。
萤火虫萤光素酶是一种分子量约为61 kDa的蛋白,在ATP、镁离子和氧气存在的条件下,可以催化luciferin氧化成oxyluciferin,在luciferin氧化的过程中,会发出生物萤光(bioluminescence)。海肾萤光素酶是一种分子量约为36 kDa的蛋白,在氧气存在的条件下,可以催化coelenterazine氧化成coelenteramide,在coelenterazine氧化过程中也会发生生物萤光。生物萤光可以通过化学发光仪(luminometer)或液闪测定仪进行测定。反应原理如下:
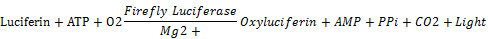
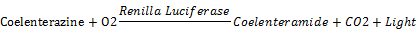
萤火虫荧光素酶催化luciferin发光的最强发光波长为560 nm,海肾萤光素酶催化coelenterazine发光的最强发光波长为465 nm。
材料(MATERIALS)
r 试剂(REAGENTS)
pNF-κB、pRenilla、过表达质粒(如果需要的话)、转染相关试剂、双萤光素酶报告基因检测试剂盒、黑底96孔板(平底)
r 试剂准备(REAGENT SETUP)
海参萤光素酶检测底物(100×)用海肾萤光素酶检测缓冲液稀释100倍至工作浓度。
如果之前样品冷冻,需要提前融化至常温。
所有试剂从冰箱拿出来解冻,并融化至常温,因为温度对于酶活有很大的影响。
r 器械(EQUIPMENT)
制冷离心机、细胞培养箱、台式小摇床、化学发光检测仪
实验步骤(PROCEDURE)
1. 细胞转染 ►预期时间消耗:3天
细胞转染详见SOP中细胞转染部分。
▲ 关键步骤:以24孔板为例,首先要设置阴性对照组pGL3和pRenilla分别加入500 ng和10 ng,再加入500ng WT的过表达质粒500 ng;然后是实验组pNF-κB和pRenilla分别加入500 ng和10 ng,再加入要过表达的质粒500 ng。
2. 双萤光素酶报告基因检测 ►预期时间消耗:1 h
3. 弃细胞培养上清,加入细胞裂解液,加入的体积如下:
器皿类型 | 96孔板 | 48孔板 | 24孔板 | 12孔板 | 6孔板 |
裂解液(μL/孔) | 100 | 150 | 200 | 300 | 500 |
4. 在台式小摇床上孵育10分钟,并以10000~15000 g速度离心3~5分钟,取上清用于测定。
5. 稀释海肾萤光素酶检测底物至工作浓度,萤火虫萤光素酶检测底物融解至常温。
6. 每个样品检测测定时,取样品20~100 μL(如果样品量足够,加入100 μL,如果样品量不足可以适当减少用量,但同一批样品的使用量宜保持一致)至黑色的平底96孔板。
7. 加入100 μL萤火虫荧光素酶检测试剂,混匀后用化学发光仪检测RLU(relative light unit)。以pGL3代替pNF-κB的那一组作为空白对照。
8. 加入100 μL海肾萤光素酶检测工作液,混匀后测定RLU。
9. 以海肾萤光素酶为内参的情况下,用萤火虫萤光素酶测定得到的RLU值除以海肾萤光素酶测定得到的RLU值。根据得到的比值来比较不同样品间目的报告基因的激活程度。
附:数据处理与结果
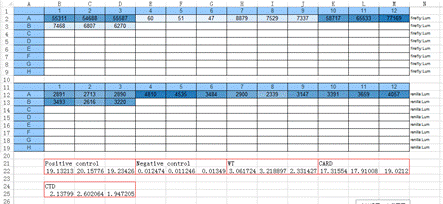
检测CARD9不同截短型激活NFκB信号通路。
1. 酶标仪检测后获得的数据如下Excel表格显示。上方为Firefly荧光素酶测定值,下方为Renilla荧光酶素测定值。下面红色方框的为对应不同实验组的Firefly荧光酶素测定值/Renilla荧光酶素测定值(各3各重复)。
2. 将求得的比值输入Prism中作图。
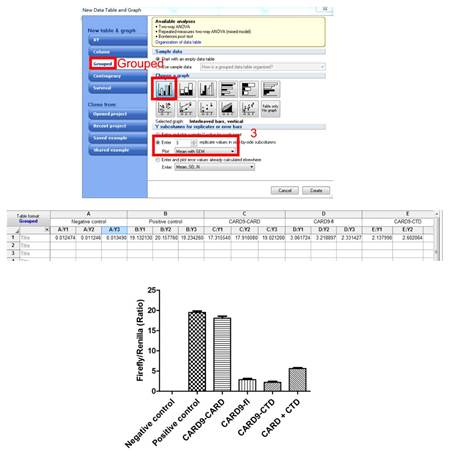
3. 利用Excel求P值(例如:CARD与fl比较)。
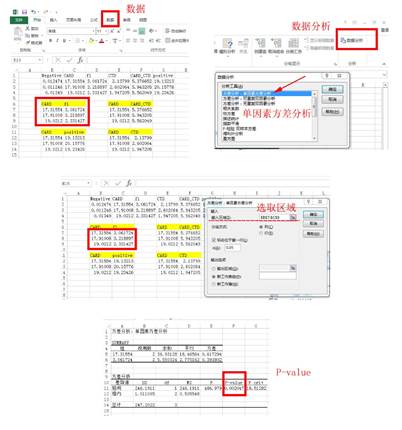
4. 在Prism中作图完成。

针对性建议(TROUBLESHOOTING)
1. 细胞裂解液可以先冻存,待以后再测定。冻存样品需要融解,并达到室温后再进行测定。
2. 加入海肾萤光素酶检测工作液后对于上一步骤中的萤火虫萤光素酶的抑制可以达到99%以上。但总会有残留微量活性,因此在转染时把海肾萤光素酶的表达量控制在其RLU读书高于萤火虫萤光素酶RLU读数的10%。
3. 检测时宜先把样品加好,并用排枪同步加入检测试剂。
4. 样品和测定试剂混合后,必须等待1~2秒,再进行测定。测定的积分时间通常为10秒,根据情况也可以修改,但是同一批样品宜使用相同的测定时间。
5. 海肾萤光素酶检测底物(100×)配制在无水乙醇中,由于无水乙醇容易挥发,有时会在初次使用时发现体积明显小于100 μL的情况,此时用无水乙醇补加至100 μL即可。
